An update version of the TOPOND program is currently available!
Release notes
TOPOND23 new features:- a complete parallelization of the main algorithms with the possibility of studying even larger and more complex systems;
- the extension of the Basis Set to the use of f and g type functions for studying the chemistry of f-electrons in complexes and compounds containing lanthanides and actinides;
- the availability of graphical tools based on Python for an accurate and flexible 2D-visualization of all calculated properties.
About the Program
The program allows for the complete and detailed topological analysis of the Electron Density, ρ(r), and its derivatives up to the fourth order, according to the Quantum Theory of Atoms in Molecules and Crystals developed by Bader.It primarily consists of:
- search and classification of charge density and Laplacian critical points (∇ ρ(r) = 0; ∇3 ρ(r) = 0);
- calculation of local quantities at the critical points: bond order, kinetic, potential and energy density, value of the virial, etc.;
- search for the atomic basins and integration of the atomic properties within them: charge, kinetic energy, virial density, Becke localization function, Shannon entropy, etc..
|
NEW!!
|
TopIso3D Viewer - A graphical tool for 3D visualization of topological properties TopIso3D Viewer is a free software with a user-friendly graphical interface, that:
|
|
The TopIso3D Viewer official web site is www.topiso3d.ufpb.br
The reference article: TopIso3D Viewer: Enhancing Topological Analysis through 3D Isosurfaces
Corresponding Author: Ary S. Maia ( arymaia@quimica.ufpb.br) NPE-LACOM, Federal University of Paraiba, Joao Pessoa, Paraiba, Brazil.
New graphical tool
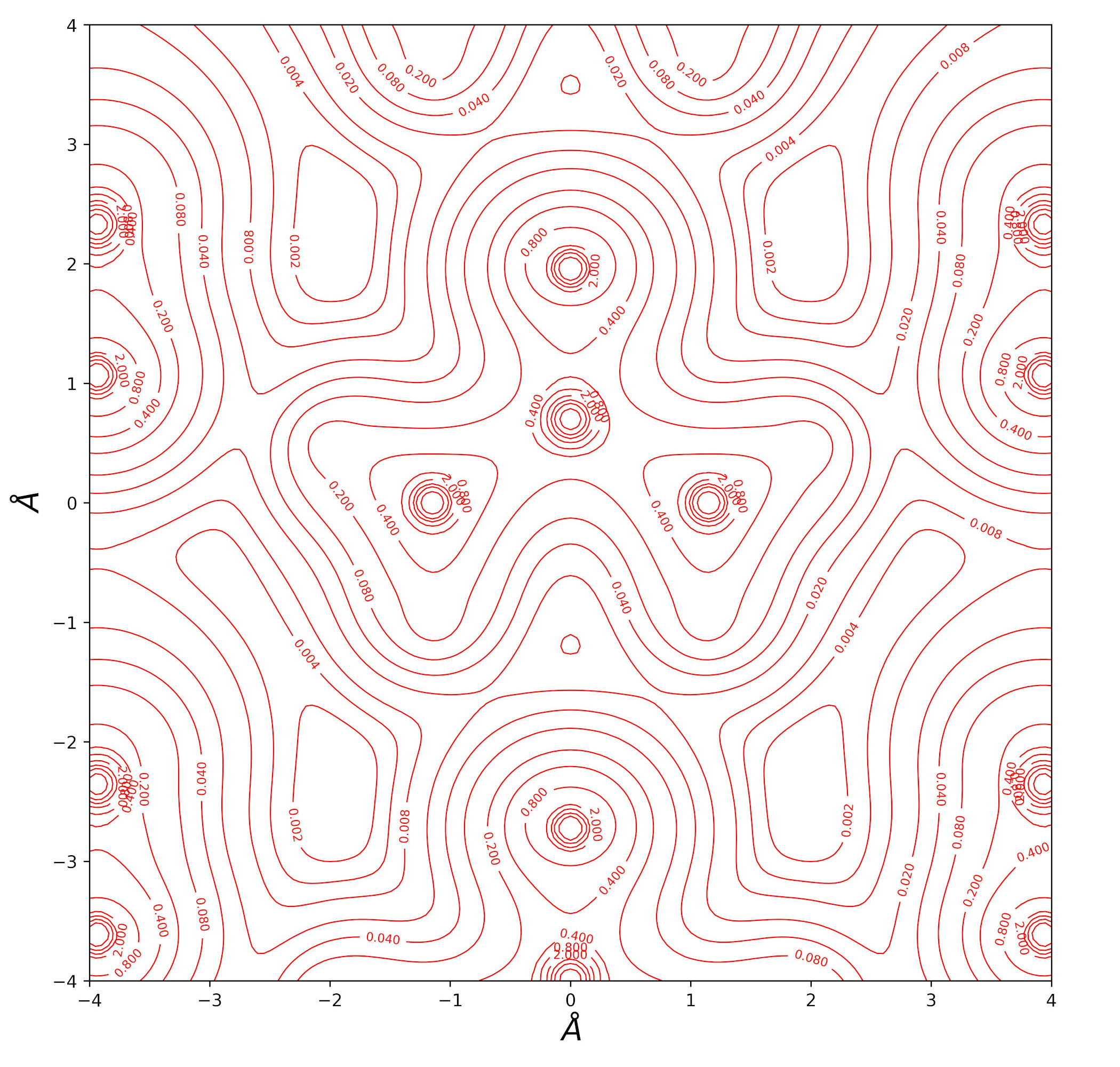
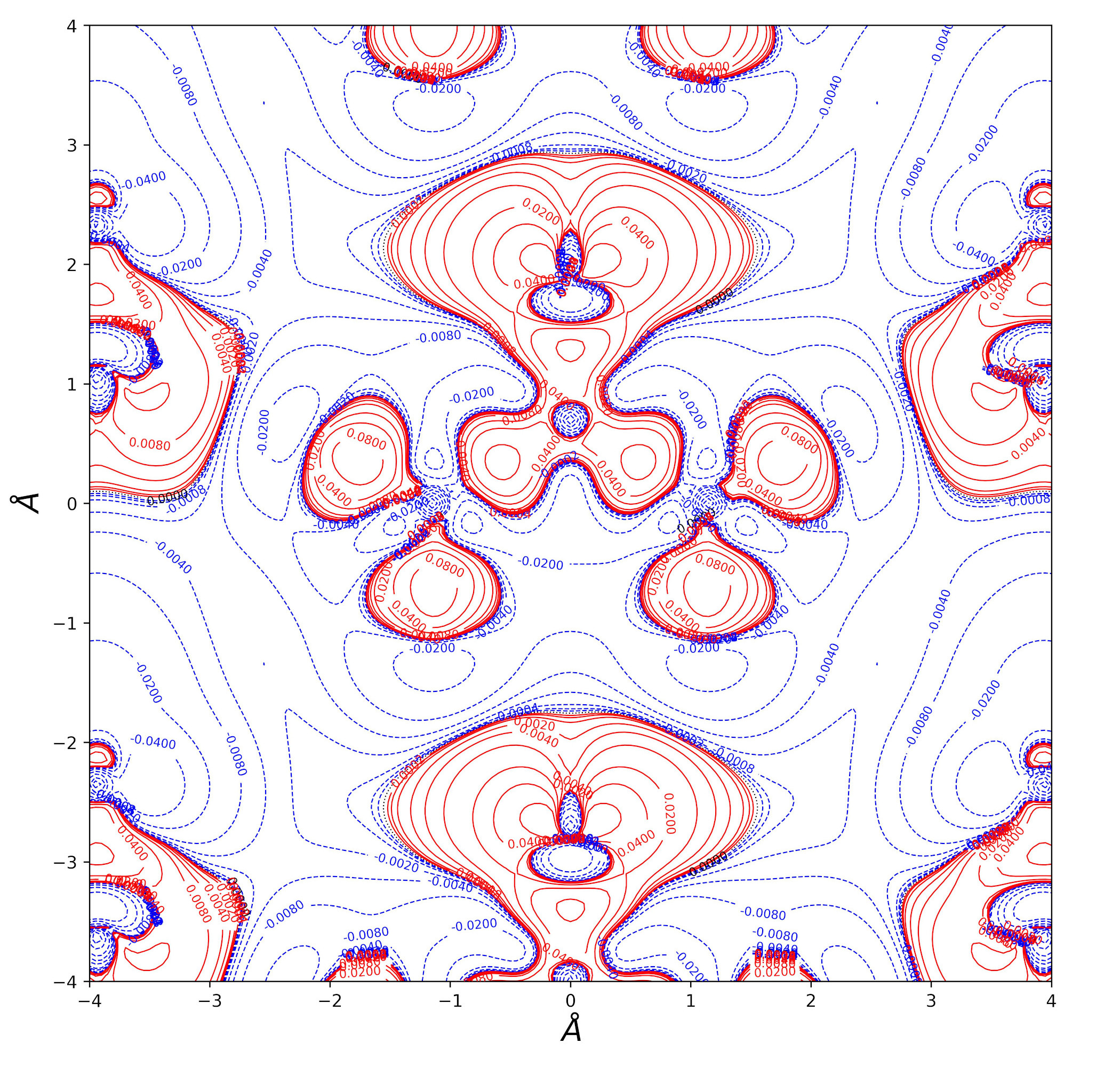
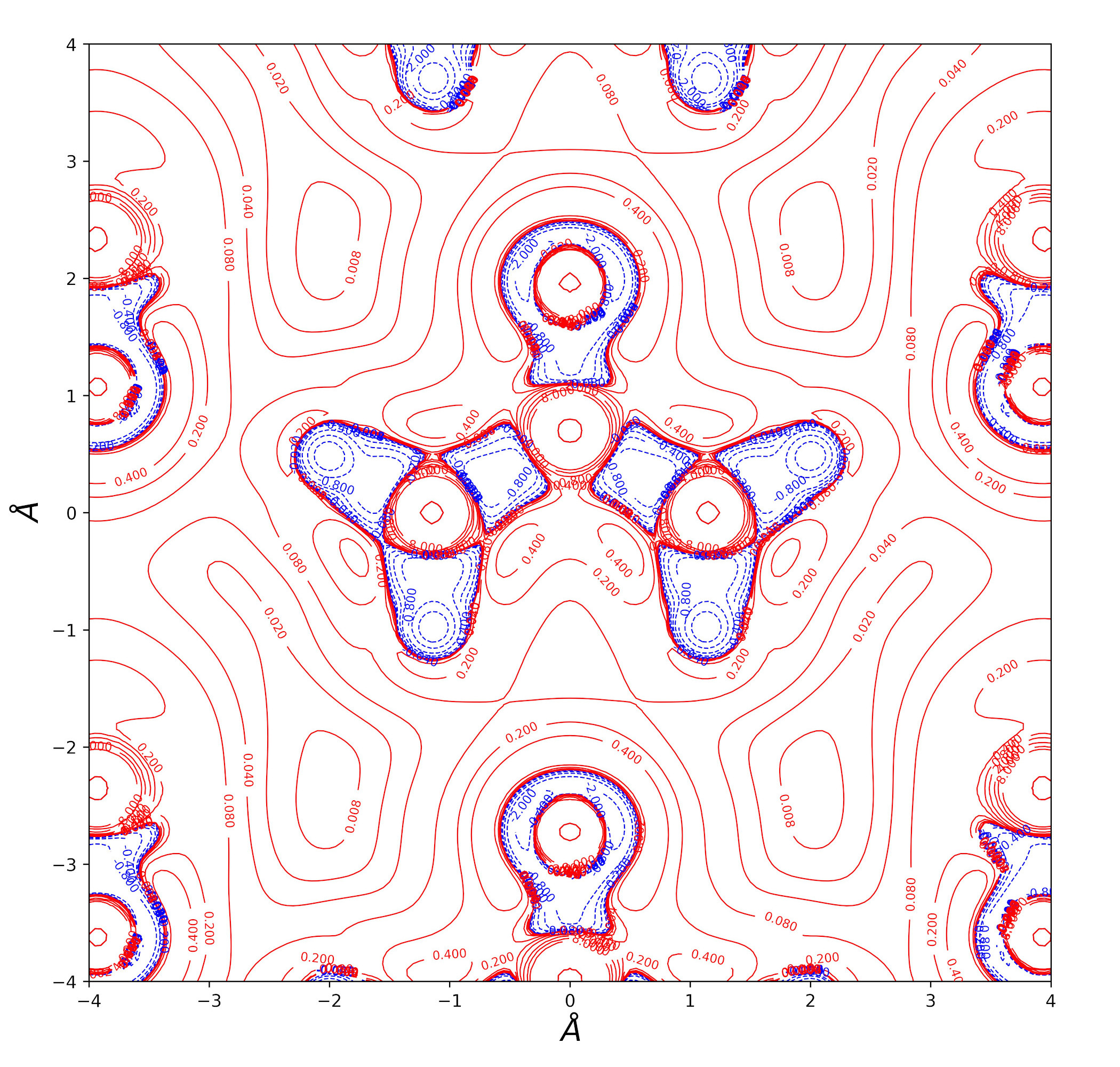
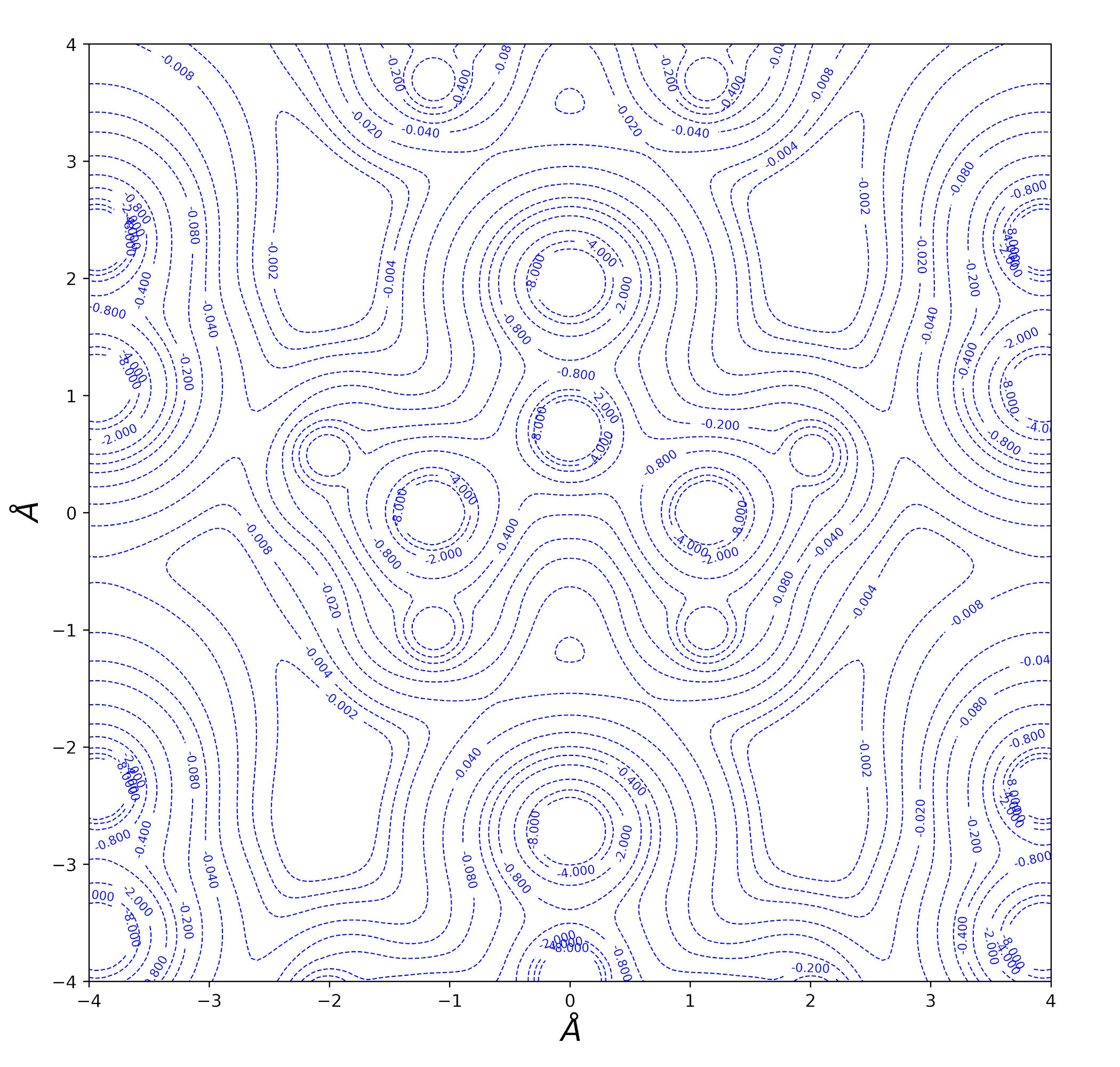
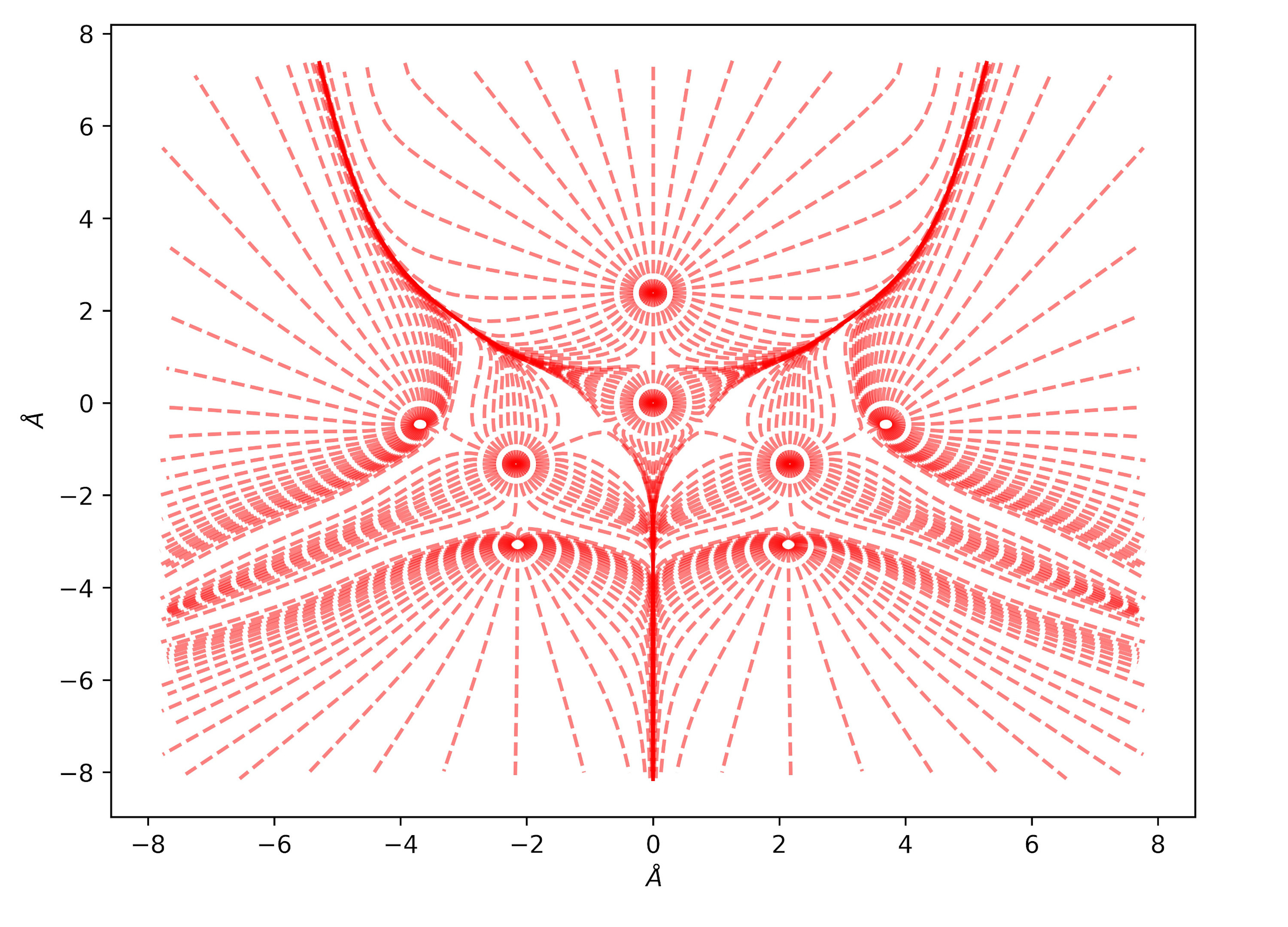
Urea bulk charge density and related topological properties with the Python based graphical tools documented in the Topond23 User's Manual.
Electron Density
Electron Density Difference
Laplacian
Virial
The trajectories of the gradient of the charge density, in the Urea molecule
The old TOPlot graphical tools are still available but not longer maintained.
A bit of history
The original TOPOND suite of programs was developed by Carlo Gatti (ISTM - CNR, Milano (Italy)) to perform the topological analysis of the Electron Density, according to the Quantum Theory of Atoms in Molecules as developed by Bader (QTAIM).Carlo Gatti proposed an extension of the method for the study of atoms in crystals and with this aim he implemented the first version of the code TOPOND98.
Since CRYSTAL14, the code has been incorporated into CRYSTAL and it is accessible by specifying the keyword TOPO in the "properties" module (see the CRYSTAL23 User's Manual for details).
While retaining all the functionalities of TOPOND98, this TOPO section of CRYSTAL enables the user to perform a topological analysis of the periodic electron density directly in CRYSTAL, thereby avoiding the use of an interface between CRYSTAL and TOPOND and the creation of external files with information from CRYSTAL.
On top of this, there is no longer the need to resize the code as a function of the dimension of the investigated system since all variables are dynamically allocated as in other parts of the CRYSTAL code.
Documentation
The following documentation is available:- Topond23 User's Manual with Python graphical tools documentation
- On-line Tutorial
- Plotting Utilities (p2dcry97.f, utilplot.f, toplot)