BARIUM (Ba Z=56)
MgO CaO SrO BaO: B1-B2 phase transition in alkaline-earth oxides,
comparison of Hartree-Fock and DF calculations [243]
The carbon analogues of type-I silicon clathrates (Ba)[118]
CaF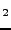 , SrF, BaF: Structural, electronic and elastic properties
[225]
, SrF, BaF: Structural, electronic and elastic properties
[225]
structural, electronic and optical properties of BaF it its cubic,
orthorhombic, and hexagonal phases [560]
Guest-framework interaction in type I inorganic clathrates with
promising thermoelectric properties: On the ionic versus neutral nature
of the alkaline-earth metal guest in A(8)Ga(16)Ge(30) (A = Sr, Ba)
[532]
SrTiO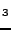 , BaTiO, PbTiO perovskites: Bulk properties and electronic
structure [450]
, BaTiO, PbTiO perovskites: Bulk properties and electronic
structure [450]
Ab initio thermodynamics of Ba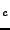 Sr
Sr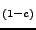 TiO3 solid solutions
[455]
TiO3 solid solutions
[455]
ABO perovskite (SrBaTiO, BaTiO, PbBaTiO) surfaces
[457] [458]
Wannier functions and chemical bonding in crystals with the perovskite-like
structure: SrTiO, BaTiO, PbTiO, and LaMnO [481]
Raman and infrared vibrational frequencies and elastic properties of solid
BaFCl calculated with various Hamiltonians: an ab initio study[213]
Vertex-linked ZnOS tetrahedra in the oxysulfide BaZnOS:
a new coordination environment for zinc in a condensed solid [529]
The performance of hybrid density functionals in solid state chemistry:
the case of BaTiO [454]
Ab initio modeling of copper adhesion on regular
BaTiO(0 0 1) surfaces [461]