


Next: About this document ...
Up: elementi
Previous: List of compounds studied
- 1
-
R. Dovesi, E. Ermondi, E. Ferrero, C. Pisani and C. Roetti, ``Hartree-Fock
study of lithium hydride with the use of a polarizable basis set'',
Phys. Rev. B 29, 3591-3600 (1984).
- 2
-
T. Asthalter, W. Weyrich, A.H. Harker, A.B. Kunz, R. Orlando and C. Pisani,
``Comparison of quasi-Hartree-Fock wave-functions for lithium hydride'',
Solid State Comm. 83, 725-730 (1992).
- 3
-
M. Causà, R. Dovesi and F. Ricca, ``Ab initio Hartree-Fock investigation of
the surface feature of LiH slabs of different thickness'',
Surf. Sci 237, 312-320 (1990).
- 4
-
I. Baraille, C. Pouchan, M. Causà and C. Pisani, ``An ab initio
Hartree-Fock study of electronic and structural properties of MgH
 '',
'',
Chem. Phys. 179, 39-46 (1994).
- 5
-
L. Ojamäe, K. Hermansson, C. Pisani, M. Causà and C. Roetti, ``Structural,
vibrational and electronic properties of a crystalline hydrate from ab initio
Hartree-Fock calculations'',
Acta Cryst. B 50, 268-279 (1994).
- 6
-
P. D'Arco, M. Causà, C. Roetti and B. Silvi, ``A periodic Hartree-Fock
study of a weakly bonded layer structure: brucite Mg(OH)
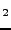 '',
'',
Phys. Rev. B 47, 3522-3529 (1993).
- 7
-
R. Dovesi, C. Pisani and C. Roetti, ``Ab initio HF versus semiempirical results
of chemisorption calculations of hydrogen on graphite'',
Chem. Phys. Lett. 81, 498-502 (1981).
- 8
-
G. Angonoa, J. Koutecky , A.N. Ermoshkin and C. Pisani, ``Ab initio
Hartree-Fock investigation of the interaction between hydrogen monolayers and
beryllium slabs'',
Surf. Sci. 138, 51-74 (1984).
- 9
-
R. Dovesi, C. Pisani, C. Roetti, J.M. Ricart and F. Illas, ``Periodic MINDO/3
study of the unreconstructed (111) surface of diamond and of hydrogen
chemisorption thereon'',
Surf. Sci. 148, 225-236 (1984).
- 10
-
R. Dovesi, C. Pisani, C. Roetti and J.M. Ricart, ``Periodic MINDO/3 study of
the reconstructed (111) surface of diamond'',
Surf. Sci. 185, 120-124 (1987).
- 11
-
A. D'Ercole, E. Giamello, C. Pisani and L. Ojamae, ``Embedded-cluster study of
hydrogen interaction with an oxygen vacancy at the MgO surface'',
J. Phys. Chem. 103, 3872-3876 (1999).
- 12
-
A. D'Ercole and C. Pisani, ``ab initio study of hydrogen dissociation at
a surface divacancy on the (001) MgO surface'',
J. Chem. Phys. 111, 9743-9753 (1999).
- 13
-
R. Dovesi, ``Ab initio Hartree-Fock approach to the study of polymers:
application to polyacetylenes'',
Int. J. Quantum Chem. 26, 197-212 (1984).
- 14
-
R. Dovesi, M. Causà, R. Orlando, C. Roetti and V.R. Saunders, ``Ab initio
approach to molecular crystals: a periodic Hartree-Fock study of crystalline
urea'',
J. Chem. Phys. 92, 7402-7411 (1990).
- 15
-
O. Maresca, R.J-M. Pellenq, F. Marinelli, J. Conard, ``A search for a strong
physisorption site for H in Li-doped porous carbon'',
J. Chem.Phys. 121, 12548-12558 (2004).
- 16
-
F.J. Torres, B. Civalleri, C. Pisani, P. Ugliengo, ``An ab initio
periodic study of acidic chabazite as a candidate for dihydrogen storage'',
J. Phys. Chem. B 110, 10467-10474 (2006).
- 17
-
R. Nada, J.B. Nicholas, M.I. McCarthy, A.C. Hess, ``Hartree-Fock studies of
zeolite/adsorbate interactions - He, Ne and Ar in silica sodalite'',
Int. J. Quantum Chem. 60, 809-820 (1996).
- 18
-
L.M. Almeida, C. Fiolhais, M. Causà, ``Properties of simple metals beyond the
local density approximation of density functional theory'',
Int. J. Quantum Chem. 91, 224-229 (2003).
- 19
-
R. Dovesi, ``Ab initio Hartree-Fock extended basis set calculation of the
electronic structure of crystalline lithium oxide'',
Solid State Comm. 54, 183-185 (1985).
- 20
-
B. Silvi, M. Causà, R. Dovesi and C. Roetti, ``Non-empirical pseudopotentials
in the HF-LCAO approach to crystalline solids. Comparison to all-electron
results'',
Molec. Phys. 67, 891-901 (1989).
- 21
-
R. Dovesi, E. Ferrero, C. Pisani and C. Roetti, ``Ab initio study of the
electron momentum distribution of metallic lithium'',
Z. Phys. B 51, 195-203 (1983).
- 22
-
K. Doll, N.M. Harrison, V.R. Saunders, ``A density functional study of lithium
bulk and surfaces'',
J. Phys. Cond. Matter 11, 5007-5019 (1999).
- 23
-
M.B. Lepetit, E. Aprà, J.P. Malrieu and R. Dovesi, ``Toward a magnetic
description of metals in terms of interstitial molecular orbitals. II.
One-dimensional infinite system: the lithium chain'',
Phys. Rev. B 46, 12974-12980 (1992).
- 24
-
W.C. Mackrodt, N.M. Harrison, V.R. Saunders, N.L. Allan and M.D. Towler,
``Direct evidence of O(P) holes in Li-doped NiO from Hartree-Fock
calculations'',
Chem. Phys. Letters 250, 66-70 (1996).
- 25
-
R. Orlando, F. Corà, R. Millini, G. Perego and R. Dovesi, ``Hydrogen
abstraction from methane by Li doped MgO: a periodic quantum mechanical
study'',
J. Chem. Phys. 105, 8937-8943 (1996).
- 26
-
A. Lichanot, C. Larrieu, R. Orlando and R. Dovesi, ``Lithium trapped-hole
centre in magnesium oxide. An ab-initio supercell study'',
J. Phys. Chem. Solids 59, 7-12 (1998).
- 27
-
D.G. Clerc and R.D. Poshusta, ``Periodic Hartree-Fock study of Li
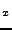 TiS
TiS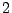 ,
,
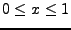 the structural, elastic, and electronic effects of lithium
intercalation in TiS'',
the structural, elastic, and electronic effects of lithium
intercalation in TiS'',
J. Phys. Chem. A 101, 8926-8931 (1997).
- 28
-
Abdurahman A, Shukla A, Dolg M, ``Ab initio treatment of electron correlations
in polymers: Lithium hydride chain and beryllium hydride polymer'',
J. Chem. Phys. 112, 4801-4805 (2000).
- 29
-
M. Merawa, P. Labeguerie, P. Ugliengo, K. Doll, R. Dovesi, ``The structural,
electronic and vibrational properties of LiOH and NaOH: an ab initio study'',
Chem. Phys. Lett. 387, 453-459 (2004).
- 30
-
R. Dovesi, C. Roetti, C. Freyria Fava, M. Prencipe and V.R. Saunders, ``On the
elastic properties of lithium, sodium an potassium oxide. An ab initio
study'',
Chem. Phys. 156, 11-19 (1991).
- 31
-
A. Lichanot, M. Gelizé, C. Larrieu and C. Pisani, ``Hartree-Fock ab initio
study of relaxation and electronic structure of lithium oxide slabs'',
J. Phys. Chem. Solids 52, 1155-1164 (1991).
- 32
-
T. Ouazzani, A. Lichanot and C. Pisani, ``Effect of the quality of the atomic
orbitals basis set about the relaxation and electronic structure of the (110)
surface of lithium oxide'',
J. Phys. Chem. Sol. 56, 915-918 (1995).
- 33
-
H. Tanigawa, M. Taniguchi, S. Tanaka, ``Interaction of hydrogen isotopes with
defects in LiO'',
Fusion Technology 34, 872-876 (1998).
- 34
-
A. Lichanot, E. Aprà and R. Dovesi, ``Quantum mechanical Hartree-Fock study
of the elastic properties of LiS and NaS'',
Phys. Stat. Sol. (b) 177, 157-163 (1993).
- 35
-
P. Azavant, A. Lichanot, M. Rerat and C. Pisani, ``Ab initio Hartree-Fock study
of lithium and sodium sulphides: electronic and scattering properties'',
Acta Cryst. B 50, 279-290 (1994).
- 36
-
A. Zupan, K. Burke, M. Ernzerhof, J.P. Perdew, ``Distributions and averages of
electron density parameters: Explaining the effects of gradient
corrections.'',
J. Chem. Phys. 106, 10184-10193 (1997).
- 37
-
T. Ouazzani, A. Lichanot, C. Pisani and C. Roetti, ``Relaxation and electronic
structure of surfaces in lithium sulphide: a Hartree-Fock ab initio
approach'',
J. Phys. Chem. Solids 54, 1603-1611 (1993).
- 38
-
A. Lichanot, M. Merawa and M. Causà, ``Density Functional LCAO calculation of
periodic systems. Effect of an ``a posteriori'' correction of the
Hartree-Fock energy on physical properties of ionic sulfur compounds'',
Chem. Phys. Letters 246, 263-268 (1995).
- 39
-
R. Dovesi, C. Pisani, F. Ricca, C. Roetti and V.R. Saunders, ``Hartree-Fock
study of crystalline lithium nitride'',
Phys. Rev. B 30, 972-979 (1984).
- 40
-
M. Causà, R. Dovesi, C. Pisani and C. Roetti, ``Ab initio study of the
autocorrelation function for lithium nitride'',
Phys. Rev. B 32, 1196-1202 (1985).
- 41
-
Schon JC; Wevers MAC; Jansen M, ``Prediction of high pressure phases in the
systems Li
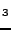 N, NaN, (Li,Na)(3)N, LiS and NaS'',
N, NaN, (Li,Na)(3)N, LiS and NaS'',
J. Mater. Chem. 11, 69-77 (2001).
- 42
-
Schon JC; Wevers MAC; Jansen M, ``Investigation of the possible ternary
nitrides in the system LiN/NaN'',
Solid St. Sciences 2, 449-456 (2000).
- 43
-
M.H. Palmer, J. A. Blair-Fish, ``Quadrupole coupling assignements in inorganic
periodic systems by ab-initio calculation of electric field gradients'',
Z. Naturforsch. 49a, 137-145 (1994).
- 44
-
R. Nada, C.R.A. Catlow, C. Pisani and R. Orlando, ``Ab initio Hartree-Fock
perturbed-cluster study of neutral defects in LiF'',
Modelling. Simul. Mater. Sci. Eng. 1, 165-187 (1993).
- 45
-
M. Causà, R. Dovesi and F. Ricca, ``Regular adsorption of CO molecules on LiF
(001)'',
Surf. Sci 280, 1-13 (1993).
- 46
-
C. Pisani, F. Corà, R. Orlando and R. Nada, ``Ab-initio perturbed-cluster
study of carbon monoxide adsorption at a stepped LiF (001) surface'',
Surf. Sci. 282, 185-194 (1993).
- 47
-
Hermansson K, Alfredsson M, ``N and HF vibrations on LiF(001): the effect
of surface coverage'',
Surf. Sci. 411, 23-34 (1998).
- 48
-
R. Nada, V.R. Saunders and C. Pisani, ``Supercell versus embedded-cluster
simulation of a lithium vacancy in a lithium fluoride monolayer'',
Chem. Phys. 169, 297-303 (1993).
- 49
-
G. Mallia, R. Orlando, C. Roetti, P. Ugliengo, R. Dovesi, ``F center in LiF: a
quantum mechanical ab initio investigation of the hyperfine interaction
between the unpaired electron and the vacancy and its first seven
neighbors'',
Phys. Rev. B 63, 235102 (2001).
- 50
-
M. Prencipe, A. Zupan, R. Dovesi, E. Aprà and V.R. Saunders, ``Ab initio
study of the structural properties of LiF, NaF, KF, LiCl, NaCl, and KCl'',
Phys. Rev. B 51, 3391-3396 (1995).
- 51
-
K. Doll and H. Stoll, ``Cohesive properties of alkali halides'',
Phys. Rev B 56, 10121-10127 (1997).
- 52
-
Ochs D, Brause M, Stracke P, Krischok S, Wiegershaus F, MausFriedrichs W,
Kempter V, Puchin VE, Shluger AL, ``The surface electronic structure of
stoichiometric and defective LiF surfaces studied with MIES and UPS in
combination with ab-initio calculations'',
Surface Sci. 383, 162-172 (1997).
- 53
-
Sims CE, Barrera GD, Allan NL, Mackrodt WC, ``Thermodynamics and mechanism of
the B1-B2 phase transition in group-I halides and group-II oxides'',
Phys. Rev. B 57, 11164-11172 (1998).
- 54
-
G. Sandrone and D.A. Dixon, ``A periodic density functional theory and
Hartree-Fock study of alkali halides with Gaussian orbitals'',
J. Phys. Chem 102, 10310-10317 (1998).
- 55
-
C. Darrigan, M. Rerat, G. Mallia, R. Dovesi, ``Implementation of the finite
field perturbation method in the CRYSTAL program for calculating the
dielectric constant of periodic systems'',
J. Comp. Chem. 24, 1305-1312 (2003).
- 56
-
M. Rerat, R. Dovesi, ``Determination of the macroscopic electric
susceptibilities
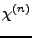 from the microscopic (hyper)polarizabilities
from the microscopic (hyper)polarizabilities
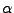 ,
, 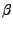 and
and 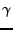 .'',
.'',
Lecture Series on Computer and Computational Sciences 1,
1-3 (2004).
- 57
-
M. Rérat, M. Ferrero, R. Dovesi, ``Evolution of the (hyper)polarizability
with the size and periodicity of the system. A model investigation from LiF
molecule to the LiF 3D crystal.'',
J. Comp. Methods in Sciences and Engineering 6, 233-242
(2006).
- 58
-
K. Doll and H. Stoll, ``Ground-state properties of heavy alkali halides'',
Phys. Rev B 57, 4327-4331 (1998).
- 59
-
W.C. Mackrodt and E.A. Williamson, ``First-principles Hartree-Fock description
of the electronic structure of monoclinic C2/m LiMnO (
 )'',
)'',
Philos. Mag B 77, 1077-1092 (1998).
- 60
-
W.C.Mackrodt, ``First principles Hartree-Fock description of lithium insertion
in oxides: I The end members TiO and LiTiO of the system
LiTiO'',
J. Solid State Chem. 142, 428-439 (1999).
- 61
-
S. Lunell, A. Stashans, L. Ojamae, H. Lindstrom, A. Hagfeldt, ``Li and Na
diffusion in TiO from quantum chemical theory versus electro chemical
experiment'',
J. Am. Chem. Soc. 119, 7374-7380 (1997).
- 62
-
A. Lichanot, C. Larrieu, C. Zicovich-Wilson, C. Roetti, R. Orlando and
R. Dovesi, ``Trapped-hole centres containing lithium and sodium in MgO, CaO
and SrO. An ab initio supercell study'',
J. Phys. Chem. Solids 59, 1119-1124 (1998).
- 63
-
N.M. Harrison, V.R. Saunders, R. Dovesi and W.C. Mackrodt, ``Transition metal
materials: a first principles approach to the electronic structure of the
insulating phase.'',
Phil. Trans. R. Soc. Lond. A 356, 75-88 (1998).
- 64
-
Alexiev V; Prins R; Weber T, ``Ab initio study of MoS and Li adsorbed on
the (10
 )0) face of MoS'',
)0) face of MoS'',
Phys. Chem. Chem. Phys. 2, 1815-1827 (2000).
- 65
-
F. Lemoigno, E. Prouzet, Z.Y. Wu, P. Gressier, G. Ouvrard, ``Combined multiple
scattering simulation and Hartree-Fock LCAO studies of atomic displacements
in LiVO
 '',
'',
Journal de Physique IV 7, 263-264 (1997).
- 66
-
B. Civalleri, A.M. Ferrari, M. Llunell, R. Orlando, M. Merawa, P. Ugliengo,
``Cation selectivity in alkali-exchanged chabazite: an ab-initio periodic
study'',
Chem. Mater. 15, 3996-4004 (2003).
- 67
-
Prencipe M, Tribaudino M, Nestola F, ``Charge-density analysis of spodumene
(LiAlSiO
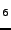 ), from ab initio Hartree-Fock calculations'',
), from ab initio Hartree-Fock calculations'',
Phys. Chem. Miner. 30, 606-614 (2003).
- 68
-
Islam MM, Maslyuk VV, Bredow T, Minot C, ``Structural and electronic properties
of LiB
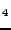 O
O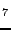 '',
'',
J. Phys. Chem. B 109, 13597-13604 (2005).
- 69
-
Bredow T, Heitjans P, Wilkening M, ``Electric field gradient calculations for
LiTiS and comparison with Li-7 NMR results'',
Phys. Rev. B 70, 115111 (2004).
- 70
-
M. Catti, ``Ab initio study of Li+ diffusion paths in the monoclinic
Li0.5CoO intercalate'',
Phys. Rev. B 61, 1795-1803 (2000).
- 71
-
Nieber H, Doll K, Zwicknagl G, ``Electronic and magnetic properties of a
hexanuclear ferric wheel'',
Eur. Phys. J. B 44, 209-215 (2005).
- 72
-
H. Nieber, K. Doll and G. Zwicknagl, ``Ab initio correlation approach to a
ferric wheel (LiFe(OCH)(12)-(dbm)(6)] PF) like molecular
cluster'',
Eur. Phys. J. B 51, 215-221 (2006).
- 73
-
A. Ramirez-Solis, C. M. Zicovich-Wilson, B. Kirtman, ``Periodic Hartree-Fock
and density functional theory calculations for Li-doped polyacetylene
chains'',
J. Chem. Phys. 124, Art. n. 244703 (2006).
- 74
-
Maslyuk VV, Islam MM, Bredow T, ``Electronic properties of compounds of the
LiO-BO system'',
Phys. Rev. B 72, Art. n. 125101 (2005).
- 75
-
Islam MM, Maslyuk VV, Bredow T, Minot C, ``Structural and electronic properties
of LiBO'',
J. Phys. Chem. B 109, 13597-13604 (2005).
- 76
-
I. V. Pentin, J. C. Schön, and M. Jansen, ``Ab initio prediction of the
low-temperature phase diagrams in the systems KBr–NaBr, KX–RbX, and
LiX–RbX (X=Cl,Br)'',
J. Chem. Phys. 126, Art. N. 124508 (2007).
- 77
-
R. Dovesi, G. Angonoa and M. Causà, ``On the core expansion of metallic
beryllium'',
Philos. Mag. 45, 601-606 (1982).
- 78
-
R. Dovesi, C. Pisani, F. Ricca and C. Roetti, ``Ab initio study of metallic
beryllium'',
Phys. Rev. B 25, 3731-3739 (1982).
- 79
-
R. Dovesi, C. Pisani, F. Ricca and C. Roetti, ``Hartree-Fock investigation of
the electron momentum distribution in metallic beryllium'',
Z. Phys. B 47, 19-26 (1982).
- 80
-
G. Angonoa, J. Koutecky and C. Pisani, ``Ab initio Hartree-Fock investigation
of beryllium slabs'',
Surf. Sci. 121, 355-370 (1982).
- 81
-
C. Freyria-Fava, R. Dovesi, V.R. Saunders, M. Leslie and C. Roetti, ``Ca and Be
substitution in bulk MgO: ab initio Hartree-Fock and ionic model supercell
calculation'',
J. Phys.: Cond. Matter 5, 4793-4804 (1993).
- 82
-
B. Soulede Bas, H.E. Dorsett, M.J. Ford, ``The electronic structure of Be
and BeO: benchmark EMS measurements and LCAO calculations'',
J. Phys. Chem. Solids 64, 495-505 (2003).
- 83
-
A. Lichanot, M. Chaillet, C. Larrieu, R. Dovesi and C. Pisani, ``Ab-initio
Hartree-Fock study of solid beryllium oxide: structure and electronic
properties'',
Chem. Phys. 164, 383-394 (1992).
- 84
-
R. Dovesi, C. Roetti, C. Freyria Fava, E. Aprà, V.R. Saunders and
N.M. Harrison, ``Ab initio Hartree-Fock treatment of ionic and semi-ionic
compounds: state of the art'',
Phil. Trans. R. Soc. Lond. A 341, 203-210 (1992).
- 85
-
Armenta MGM; Reyes-Serrato A; Borja MA, ``Ab initio determination of the
electronic structure of beryllium-, aluminum-, and magnesium-nitrides: A
comparative study'',
Phys. Rev. B 62, 4890-4898 (2000).
- 86
-
Ma. Guadalupe Moreno Armanta, Armando Reyes-Serrato, ``Direct wide gap
material: a Hartree-Fock study of -BeN'',
Comp. Mater. Science 21, 95-100 (2001).
- 87
-
Y. Noel, C.M. Zicovich-Wilson, B. Civalleri, Ph. D'Arco, R. Dovesi,
``Polarization properties of ZnO and BeO: an ab-initio study through the
Berry phase and Wannier functions approaches'',
Phys. Rev. B 64, 014111 (2002).
- 88
-
M. Merawa, M. Llunell, R. Orlando, M. Gelize-Duvignau, R. Dovesi, ``Structural,
electronic and elastic properties of some fluoride crystals: an ab initio
study'',
Chem. Phys. Letters 368, 7-11 (2003).
- 89
-
M. Prencipe, ``Ab initio Hartree-Fock study and charge density analysis of
beryl (AlBeSi
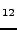 O
O )'',
)'',
Phys. Chem. Minerals 29, 552-561 (2002).
- 90
-
R. Dovesi, C. Pisani and C. Roetti, ``Exact exchange Hartree-Fock calculations
for periodic systems. II. Results for graphite and hexagonal boron nitride'',
Int. J. Quantum Chem. 17, 517-529 (1980).
- 91
-
R. Dovesi, C. Pisani, C. Roetti and P. Dellarole, ``Exact exchange Hartree-Fock
calculations for periodic systems. IV. Ground state properties of cubic boron
nitride'',
Phys. Rev. B 24, 4170-4176 (1981).
- 92
-
A. Lichanot, M. Rerat and M. Causà, ``LCAO-LDA calculation of Compton
profiles in hexagonal BN: Comparison with experiments.'',
J. Phys.: Cond. Matter 8, 10425-10434 (1996).
- 93
-
D. Ayma, M. Rerat, A. Lichanot, ``Ab initio self-consistent calculations of the
Compton profiles and polarizabilities of diamond and cubic boron nitride'',
J. Phys. Cond. Matter 10, 557-575 (1998).
- 94
-
T. Asthalter, M. Walter, ``Visualizing covalency and multiple bonds in terms of
the electronic single-particle density matrix'',
Z. Phys. Chem 215, 1277-1288 (2001).
- 95
-
M. Causà, R. Dovesi and C. Roetti, ``Pseudopotential Hartree-Fock study of
seventeen III-V and IV-IV semiconductors'',
Phys. Rev. B 43, 11937-11943 (1991).
- 96
-
M. Causà and A. Zupan, ``Density Functional LCAO calculations for solids:
comparisons between Hartree-Fock and Kohn-Sham structural properties'',
Int. J. Quantum Chem. QCS 28, 633-644 (1994).
- 97
-
A. Zupan and M. Causà, ``Density Functional LCAO calculations for solids.
Comparison among Hartree-Fock, DFT Local Density Approximation, and DFT
Generalized Gradient Approximation structural properties'',
Int. J. Quantum Chem. 56, 337-344 (1995).
- 98
-
Perottoni CA; Pereira AS; da Jornada JAH, ``Periodic Hartree-Fock linear
combination of crystalline orbitals calculation of the structure, equation of
state and elastic properties of titanium diboride'',
J. Phys. - Cond. Matter 12, 7205-7222 (2000).
- 99
-
Zapol P; Curtiss LA; Erdemir A, ``Periodic ab initio calculations of orthoboric
acid'',
J. Chem. Phys. 113, 3338-3343 (2000).
- 100
-
V. Luãna, A. Costales, P. Mori-Sánchez, A. Martin Pendás, ``Ions
in crystals: The topology of the Electron density in ionic materials. 4. The
danburite (CaBSiO
 ) case of the occurrence of oxide-oxide bond
paths in crystals.'',
) case of the occurrence of oxide-oxide bond
paths in crystals.'',
J. Phys. Chem. B 107, 4912-4921 (2003).
- 101
-
Larina AV, Vercauteren DP, ``Comparison of small size alumino- and
borosilicates optimised by periodic Hartree-Fock'',
Stud. Surf. Sci. Catal. 142, 1987-1994 (2002).
- 102
-
M.M. Islam, T. Bredow, C. Minot, ``Comparison of trigonal BO structures
with high and low space-group symmetry'',
Chem. Phys. Letters 418, 565-568 (2006).
- 103
-
Deak P, Gali A, Solyom A, Buruzs A, Frauenheim T, ``Electronic structure of
boron-interstitial clusters in silicon'',
JOURNAL OF PHYSICS-CONDENSED MATTER 17, S2141-S2153
(2005).
- 104
-
R. Dovesi, C. Pisani, F. Ricca and C. Roetti, ``Exact exchange Hartree-Fock
calculations for periodic systems. III. Ground state properties of diamond'',
Phys. Rev. B 22, 5936-5944 (1980).
- 105
-
D. Ayma, J.P. Campillo, M. Rerat and M. Causà, ``Parallel Computation of the
dynamic polarizability and dielectric constant of the Carbon and Silicon
cubic crystals'',
J. of Comp. Chem. 18, 1253-1263 (1997).
- 106
-
K.H. Lee, M. Causà, S.S. Park, ``Ab initio Periodic Hartree-Fock calculations
for interpretation of the scanning tunneling microscope (STM) images of
graphite'',
J. Phys. Chem. B 102, 6020-6024 (1998).
- 107
-
L. Pisani, J.A. Chan, B. Montanari, N.M. Harrison, ``Electronic structure and
magnetic properties of graphitic ribbons'',
Phys. Rev. B 75, Art. n. 064418 (2007).
- 108
-
C. Pisani, R. Orlando and R. Nada, ``Ab initio Hartree-Fock perturbed-cluster
treatment of local defects in crystals: application to carbon impurities in
silicon'',
Rev. Solid State Sci. 5, 177-196 (1991).
- 109
-
R. Orlando, R. Dovesi, P. Azavant, N.M. Harrison and V.R. Saunders, ``A
super-cell approach for the study of localized defects in solids: carbon
substitution in bulk silicon'',
J. Phys.: Cond. Matter 6, 8573-8583 (1994).
- 110
-
R. Orlando, P. Azavant, M.D. Towler, R. Dovesi and C. Roetti, ``Cluster and
supercell calculations for carbon-doped silicon'',
J. Phys.: Cond. Matter 8, 1123-1133 (1996).
- 111
-
R. Dovesi and R. Orlando, ``Convergence properties of the supercell approach in
the study of local defects in solids'',
Phase Trans. 52, 151-167 (1994).
- 112
-
M. Catti,A. Pavese, R. Dovesi and V. R. Saunders, ``Static lattice and electron
properties of MgCO (magnesite) calculated by ab initio periodic
Hartree-Fock methods'',
Phys. Rev. B 47, 9189-9198 (1993).
- 113
-
M. Catti, A. Pavese, E. Aprà and C. Roetti, ``Quantum-mechanical
Hartree-Fock study of calcite (CaCO) at variable pressure, and comparison
with magnesite (MgCO)'',
Phys. Chem. Minerals 20, 104-110 (1993).
- 114
-
Gali A, Deak P, Son NT, Janzen E, ``Possibility for the electrical activation
of the carbon antisite by hydrogen in SiC'',
Phys. Rev. B 71, Art. N. 035213 (2005).
- 115
-
Mori-Sanchez P, Marques M, Beltran A, Jiang JZ, Gerward L, Recio JM, ``Origin
of the low compressibility in hard nitride spinels'',
Phys. Rev. B 68, art no 064115 (2003).
- 116
-
M. Catti, ``Orthorhombic intermediate state in the zinc blende to rocksalt
transformation path of SiC at high pressure'',
Phys. Rev. Letters 87, art 035504 (2001).
- 117
-
A. Gali, P. Deak, E. Rauls, N.T. Son, I.G. Ivanov, F.H.C. Carlsson, E. Janzen,
W.J. Choyke, ``Correlation between the antisite pair and the
 center in
SiC'',
center in
SiC'',
Phys. Rev. B 67, Art N. 155203 (2003).
- 118
-
C.A. Perottoni and A.H. de Jornada, ``The carbon analogues of type-I silicon
clathrates'',
J. Phys: Condens. Matter 13, 5981-5998 (2001).
- 119
-
M. Milanesio, R. Bianchi, P. Ugliengo, C. Roetti and D. Viterbo, ``Vitamin C at
120 K: experimental and theoretical study of the charge density'',
J. Mol. Structure THEOCHEM 419, 139-154 (1997).
- 120
-
Starikov EB, ``Hartree-Fock crystal orbital calculation on sodium-intercalated
fullerites C
 Na
Na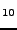 and CNa
and CNa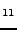 '',
'',
Chem. Phys. 256, 149-158 (2000).
- 121
-
Chan JA, Montanari B, Gale JD, Bennington SM, Taylor JW, Harrison NM,
``Magnetic properties of polymerized C-60: The influence of defects and
hydrogen'',
Phys. Rev. B 70, Art 041403 (2004).
- 122
-
Knaup JM, Deak P, Frauenheim T, Gali A, Hajnal Z, Choyke WJ, ``Theoretical
study of the mechanism of dry oxidation of 4H-SiC'',
Phys. Rev. B 71, Art. N. 235321 (2005).
- 123
-
R. Pandey, M. Causà, N.M. Harrison and M. Seel, ``The high pressure phase
transition of silicon and gallium nitride: a comparative study of
Hartree-Fock and Density Functionals calculations'',
J. Phys.: Cond. Matter. 8, 3993-4000 (1996).
- 124
-
Jiang JZ, Lindelov H, Gerward L, Stahl K, Recio JM, Mori-Sanchez P, Carlson S,
Mezouar M, Dooryhee E, Fitch A, Frost DJ, ``Compressibility and thermal
expansion of cubic silicon nitride'',
Phys. Rev. B 65, - (2002).
- 125
-
V.M. Bermudez, ``Theoretical study of the electronic structure of the
SiN(0001) surface'',
Surface Sci. 579, 11-20 (2005).
- 126
-
Paszkowicz W, Minikayev R, Piszora P, Knapp M, Bahtz C, Recio JM, Marques M,
Mori-Sanchez P, Gerward L, Jiang JZ, ``Thermal expansion of spinel-type
SiN'',
Phys. Rev. B 69, art no 052103 (2004).
- 127
-
I. de P.R. Moreira, R. Dovesi, ``Periodic approach to the electronic structure
and magnetic coupling in KCuF, KCuF, and SrCuOCl
low-dimensional magnetic systems'',
Int. J. Quantum Chem 99, 805-823 (2004).
- 128
-
Hess WP, German KAH, Bradley RA, McCarthy M, ``Laser ablation of sodium nitrate
- NO desorption following excitation of the
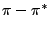 band of the nitrate
anion'',
band of the nitrate
anion'',
Applied Surface Sci. 96, 321-325 (1996).
- 129
-
McCarthy MI, Peterson KA, Hess WP, ``Electronic structure of sodium nitrate -
investigations of laser-desorption mechanisms'',
J. Phys. Chem. 100, 6708-6714 (1996).
- 130
-
R. Dovesi, C. Pisani, C. Roetti and V.R. Saunders, ``Hartree-Fock study of
polysulphur-nitride. I. The isolated infinite chain'',
J. Chem. Phys. 81, 2839-2844 (1984).
- 131
-
M. Causà, R. Dovesi, C. Pisani, C. Roetti and V.R. Saunders, ``Hartree-Fock
study of polysulphur nitride. II. Three-dimensional structures and interchain
interactions'',
J. Chem. Phys. 88, 3196-3203 (1988).
- 132
-
C. Gatti, V.R. Saunders and C. Roetti, ``Crystal field effects on the
topological properties of the electron density in molecular crystals. The
case of urea'',
J. Chem. Phys. 101, 10686-10696 (1994).
- 133
-
M.H. Palmer, J. A. Blair-Fish, ``Quadrupole coupling assignements in some
H-bonded organic systems by ab-initio lattice calculations of electric field
gradients'',
Z. Naturforsch. 49a, 146-154 (1994).
- 134
-
R. Weihrich, S. F. Matar, E. Betranhandy, V. Eyert, ``A model study for the
breaking of N2 from CNx within DFT.'',
Solid State Sci. 5, 701-703 (2003).
- 135
-
T. Bredow, ``Theoretical investigation of nitrogen substitution in cubic
zirconia'',
Phys. Rev. B 75, Art. N. 144102 (2007).
- 136
-
M. Causà, R. Dovesi, C. Pisani and C. Roetti, ``Electron charge density and
electron momentum distribution in magnesium oxide'',
Acta Cryst. B 42, 247-253 (1986).
- 137
-
M. Causà, R. Dovesi, C. Pisani and C. Roetti, ``Electronic structure and
stability of different crystal phases of magnesium oxide'',
Phys. Rev. B 33, 1308-1316 (1986).
- 138
-
M. Causà, R. Dovesi, C. Pisani and C. Roetti, ``Ab initio Hartree-Fock study
of the MgO (001) surface'',
Surf. Sci. 175, 551-560 (1986).
- 139
-
M. Causà, R. Dovesi, C. Pisani and C. Roetti, ``Directional Compton profiles
and autocorrelation function of magnesium oxide'',
Phys. Rev. B 34, 2939-2941 (1986).
- 140
-
R. Orlando, R. Dovesi, C. Roetti and V.R. Saunders, ``Convergence properties of
the cluster model in the study of local perturbations in ionic systems. The
case of bulk defects in MgO'',
Chem. Phys. Lett. 228, 225-232 (1994).
- 141
-
R. Nada, A.C. Hess and C. Pisani, ``Topological defects at the (001) surface of
MgO: energetics and reactivity'',
Surf. Sci. 336, 353-361 (1995).
- 142
-
U. Birkenheuer, F. Corà, C. Pisani, I. Scorza and G. Perego,
``Embedded-cluster study of core-level binding energies of magnesium and
alkali impurities at the surface of MgO'',
Surf. Sci. 373, 393-408 (1997).
- 143
-
R. Orlando, R. Millini, G. Perego and R. Dovesi, ``Catalytic properties of the
F-centres at the magnesium oxide surface'',
J. Molec. Catal.A; Chem 119, 253-262 (1997).
- 144
-
Ph. D'Arco, G. Sandrone, R. Dovesi, E. Aprà and V.R. Saunders, ``A
quantum-mechanical study of the relative stability under pressure of
MgSiO-ilmenite, MgSiO-perovskite, and MgO-periclase +
SiO-stishovite assemblage'',
Chem. Phys. Minerals 21, 285-293 (1994).
- 145
-
C. Pisani, R. Dovesi, R. Nada and S. Tamiro, ``Ab initio Hartree-Fock
perturbed-cluster treatment of local chemisorption. Isolated carbon monoxide
on a periodic MgO (100) substrate'',
Surf. Sci 216, 489-504 (1989).
- 146
-
M. Catti, G. Valerio, R. Dovesi and M. Causà, ``Quantum-mechanical
calculations of the solid-state equilibrium MgO + -AlO
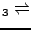 MgAlO (spinel) versus pressure'',
MgAlO (spinel) versus pressure'',
Phys. Rev. B 49, 14179-14187 (1994).
- 147
-
M.I. McCarthy and N.M. Harrison, ``Ab Initio determination of the bulk
properties of MgO.'',
Phys. Rev. B 49, 8574-8582 (1994).
- 148
-
E. Heifets, E. A. Kotomin and R. Orlando, ``Periodic Hartree-Fock simulation of
the Ag/MgO interface structure'',
J. Phys. Condens. Matter 8, 6577-6584 (1996).
- 149
-
E. Heifets, Y.F. Zhukovskii, E.A. Kotomin, M. Causà, ``The adhesion nature of
the Ag/MgO (100) interface: an ab initio study'',
Chem. Phys. Lett. 238, 395-401 (1998).
- 150
-
K.D. Heath, W.C. Mackrodt, V.R. Saunders and M. Causà, ``Calculated
enthalpies of mixing of MnO/MgO and NiO/MgO'',
J. Mater. Chem. 4, 825-829 (1994).
- 151
-
M.D. Towler, N.L. Allan, N.M. Harrison, V.R. Saunders, W.C. Mackrodt,
``Localized electron behavior within band theory - A Hartree-Fock description
of M Mg
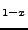 O (M=Mn, Ni)'',
O (M=Mn, Ni)'',
J. Phys. Cond. Matter 7, 6231-6239 (1995).
- 152
-
M.D. Towler, ``Ab Initio study of the surface and interfacial properties of
layered MgO/NiO film.'',
Phys. Rev. B 52, 5375-5384 (1995).
- 153
-
M.D. Towler, N.M. Harrison and M.I. McCarthy, ``Ab initio study of the surface
and interfacial properties of a layered MgO/NiO film'',
Phys. Rev. B 52, 5375-5384 (1995).
- 154
-
J.E. Jaffe, N.M. Harrison and A.C. Hess, ``Ab initio study of ZnO (1010)
surface relaxation.'',
Phys. Rev. B 49, 11153-11158 (1994).
- 155
-
M. Causà, R. Dovesi, E. Kotomin and C. Pisani, ``MgO (110) surface and CO
adsorption thereon. I. Clean (110) surface'',
J. Phys. C: Solid State Phys. 20, 4983-4990 (1987).
- 156
-
M. Causà, E. Kotomin, C. Pisani and C. Roetti, ``MgO (110) surface and CO
adsorption thereon. II. CO adsorption'',
J. Phys. C: Solid State Phys. 20, 4991-4997 (1987).
- 157
-
R. Dovesi, R. Orlando, F. Ricca and C. Roetti, ``CO adsorption on MgO crystals:
Hartree-Fock calculations for regular adlayers on a (001) lattice plane'',
Surf. Sci. 186, 267-278 (1987).
- 158
-
Y. Ferro, A. Allouche, F. Corà, C. Pisani and C. Girardet, ``Adsorption of
NH
 on MgO (100): a comparative study of ab initio and semi-classical
calculations'',
on MgO (100): a comparative study of ab initio and semi-classical
calculations'',
Surf. Science 325, 139-150 (1995).
- 159
-
M. Causà, R. Dovesi, C. Roetti, E. Kotomin and V.R. Saunders, ``A periodic ab
initio Hartree-Fock calculation on corundum'',
Chem. Phys. Letters 140, 120-123 (1987).
- 160
-
C. Pisani, M. Causà, R. Dovesi and C. Roetti, ``Hartree-Fock ab initio
characterization of ionic crystal surfaces with a slab model. The (0001) face
of -AlO'',
Progr. in Surf. Sci 25, 119-137 (1987).
- 161
-
M. Causà, R. Dovesi, C. Pisani and C. Roetti, ``Ab initio characterization of
the (0001) and (100) crystal faces of - alumina'',
Surf. Sci 215, 259-271 (1988).
- 162
-
L. Salasco, R. Dovesi, R. Orlando, C. Pisani, M. Causà and V.R. Saunders, ``A
Periodic ab initio extended basis set study of -AlO'',
Mol. Phys. 72, 267-277 (1992).
- 163
-
R. Dovesi, C. Roetti, M. Causà and C. Pisani, Ab initio study of the
periodic carbon monoxide adsorption on the basal plane of - alumina,
in Structure and Reactivity of Surfaces, edited by C. Morterra, A.
Zecchina and G. Costa, 385-393, Elsevier Science, Amsterdam (1989).
- 164
-
McCarthy MI, Schenker GK, Scamehorn CA, Nicholas JB, ``Structure and dynamics
of the water/MgO interface'',
J. Phys. Chem 100, 16989-16995 (1996).
- 165
-
McCarthy MI, Schenter GK, Chacon-Taylor MR, Rehr JJ, Brown GE, ``Prediction of
extended x-ray absorption fine structure spectra from molecular interaction
models: Na+(H2O)(n)-MgO(100) interface'',
Phys. Rev. B 56, 9925-9936 (1997).
- 166
-
Scamehorn CA, Harrison NM, McCarthy MI, ``Water chemistry on surface defect
sites - Chemidissociation versus physisorption on MgO(001)'',
J. Chem. Phys. 98, 6387-6391 (1993).
- 167
-
R. Dovesi, C. Pisani, C. Roetti and B. Silvi, ``The electronic structure of
-quartz: a periodic Hartree-Fock calculation'',
J. Chem. Phys 86, 6967-6971 (1987).
- 168
-
R. Nada, C.R.A. Catlow, R. Dovesi and C. Pisani, ``An Ab-Initio Hartree-Fock
Study of -Quartz and Stishovite'',
Phys. and Chem. Minerals 17, 353-362 (1990).
- 169
-
B. Silvi, P. D'Arco and M. Causà, ``Periodic pseudopotential Hartree Fock
study of the -quartz structure of SiO and GeO'',
J. Chem. Phys. 93, 7225-7229 (1990).
- 170
-
B. Silvi, P. D'Arco, V.R. Saunders and R. Dovesi, ``Periodic Hartree-Fock study
of minerals: tetracoordinate silica polymorphs'',
Phys. Chem. Minerals 17, 674-680 (1991).
- 171
-
F. Corà and C. Pisani, ``A quantum-mechanical ab-initio simulation of
neutral and charged point defects in alpha-quartz'',
Modelling. Simul. Mater. Sci. Eng. 2, 965-974 (1994).
- 172
-
V.B. Sulimov, C. Pisani, F. Corà and V.O. Sokolov, ``Isolated and embedded
cluster modelling of the oxygen vacancy in -quartz'',
Comp. Phys. Comm. 90, 511-514 (1994).
- 173
-
W.C. Mackrodt, N.M Harrison, V.R. Saunders, N.L.Allan, M.D. Towler, E. Aprà
and R. Dovesi, ``Ab initio Hartree-Fock calculations of CaO, VO, MnO and
NiO'',
Philos. Magaz. A 68, 653-666 (1993).
- 174
-
G. Bussolin, S. Casassa, C. Pisani and P. Ugliengo, ``Ab Initio of HCl and HF
interaction with crystalline ice. I. Physical adsorption'',
J. Chem. Phys. 108, 9516-9528 (1998).
- 175
-
A. Fahmi, C. Minot, B. Silvi and M. Causà, ``Theoretical analysis of the
structures of titanium dioxide crystals'',
Phys. Rev. B 47, 11717-11724 (1993).
- 176
-
P. Reinhardt, B.A. Hess and M. Causà, ``Electronic and geometrical structure
of bulk rutile studied with Hartree-Fock and Density Functional methods'',
Int. J. Quantum Chem. 58, 297-306 (1996).
- 177
-
M. Catti, G. Sandrone and R. Dovesi, ``Periodic Unrestricted Hartree-Fock study
of corundum-like TiO and VO'',
Phys. Rev B 55, 16122-16131 (1997).
- 178
-
W.C. Mackrodt, H.J. Gotsis and N.L. Allan, ``First principles calculations of
solute-hole interactions in the high T
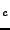 oxyfluoride superconductor Sr
CuOF'',
oxyfluoride superconductor Sr
CuOF'',
Ber.Bunsenges. Phys. Chem. Chem. Phys. 101, 1242-1244
(1997).
- 179
-
E. Ruiz, S. Alvarez, P. Alemany, R.A. Evarestov, ``Electronic structure and
properties of CuO'',
Phys. Rev B 56, 7189-7196 (1997).
- 180
-
P. Reinhardt, M. Causà, C.M. Marian and B.A. Heß, ``Adsorption of CO on
TiO (110) studied by means of a cluster model surrounded with fitted
multipoles in comparison to a periodic Ansatz'',
Phys. Rev. B 54, 14812-14820 (1996).
- 181
-
M. Catti, G. Sandrone, G. Valerio and R. Dovesi, ``Electronic, magnetic and
crystal structure of CrO by theoretical methods'',
J. Phys. Chem. Solids 57, 1735-1741 (1996).
- 182
-
M.D. Towler, N.L. Allan, N.M. Harrison, V.R. Saunders, W.C. Mackrodt and E.
Aprà, ``An ab initio Hartree-Fock study of MnO and NiO'',
Phys. Rev. B 50, 5041-5054 (1994).
- 183
-
F. Freyria Fava, I. Baraille, A. Lichanot, C. Larrieu and R. Dovesi, ``On the
structural, electronic and magnetic properties of MnCrO spinel'',
J. Phys.: Cond. Matter 9, 10715-10724 (1997).
- 184
-
M. Catti, G. Valerio and R. Dovesi, ``Theoretical study of electronic,
magnetic, and structural properties of - FeO (hematite)'',
Phys. Rev. B 51, 7441-7450 (1995).
- 185
-
W.C. Mackrodt, F. Jollet and M. Gautier-Soyer, ``A first-principles
Hartree-Fock interpretation of the X-ray oxygen K- edge spectrum of hematite
(alpha-FeO'',
Phil. Mag B 79, 25-36 (1999).
- 186
-
P. Azavant, A. Lichanot, ``X-ray scattering factors of oxygen and sulfur ions:
an ab-initio Hartree-Fock calculation'',
Acta Cryst. A 49, 91-97 (1993).
- 187
-
R. Pandey, J.E. Jaffe, N.M. Harrison, ``Ab initio study of high-pressure phase
transition in GaN'',
J. Phys. Chem. Solids 55, 1357-1361 (1994).
- 188
-
J.M. Recio, M.A. Blanco, V. Luana, R. Pandey, L. Gerward, J.S. Olsen,
``Compressibility of the high-pressure rocksalt phase of ZnO'',
Phys. Rev B 58, 8949-8954 (1998).
- 189
-
R. Tappero and A. Lichanot, ``A comparative study of the electronic structure
of alpha-MnS (alabandite) calculated at the Hartree-Fock and density
functional levels of theory'',
Chem. Phys. 236, 97-105 (1998).
- 190
-
A. Zupan, I. Petek, M. Causà and R. Dovesi, ``Elastic constants, phase
transition and electronic structure of strontium oxide (SrO): an ab initio
Hartree-Fock study'',
Phys. Rev. B 48, 799-806 (1993).
- 191
-
R. Orlando, C. Pisani, C. Roetti and E. Stefanovich, ``Ab initio Hartree-Fock
study of tetragonal and cubic phases of zirconium dioxide'',
Phys. Rev. B 45, 592-601 (1992).
- 192
-
R. Orlando, C. Pisani, E. Ruiz and P. Sautet, ``Ab-initio study of the bare and
hydrated (001) surface of tetragonal zirconia'',
Surf. Sci. 275, 482-492 (1992).
- 193
-
R. Orlando, C. Pisani, C. Roetti and E. Stefanovich, Ab initio
Hartree-Fock study of tetragonal and cubic phases of zirconium dioxide, In
Defects in insulating materials, volume 1, 630-632, World Scientific,
Schloß Nordkirchen, Germany (1992).
- 194
-
R.A. Evarestov, A.V. Leko, V.A. Veryazov, ``Hartree-Fock study of the chemical
bonding in crystalline titanium oxides: TiO, TiO, TiO'',
Phys. Status Solidi R3, 203- (1997).
- 195
-
F. Corà, A. Patel, N. M. Harrison, R. Dovesi and C.R.A. Catlow, ``An
ab-initio Hartree-Fock study of the cubic and tetragonal phases of bulk
tungsten trioxide, WO'',
J. Am Chem. Soc. 118, 12174-12182 (1996).
- 196
-
R. Dovesi, C. Pisani, C. Roetti, M. Causà and V.R. Saunders,
CRYSTAL88, An ab initio all-electron LCAO-Hartree-Fock program
for periodic systems. QCPE Pgm N.577,
Quantum Chemistry Program Exchange, Indiana University, Bloomington,
Indiana (1989).
- 197
-
Ph. D'Arco, G. Sandrone, R. Dovesi, R. Orlando and V.R. Saunders, ``A quantum
mechanical study of the perovskite structure type of MgSiO'',
Phys. Chem. Minerals 20, 407-414 (1993).
- 198
-
R. Nada, C.R.A. Catlow, R. Dovesi and V.R. Saunders, ``An ab initio
Hartree-Fock study of the ilmenite-structured MgSiO'',
Proc. Royal Soc. Lond. A 436, 499-509 (1992).
- 199
-
P. D'Arco, B. Silvi, C. Roetti and R. Orlando, ``Comparative study of spinel
compounds: a pseudopotential periodic HF calculation of MgSiO
 ,
MgGeO, AlMgO and GaMgO'',
,
MgGeO, AlMgO and GaMgO'',
J. Geophys. Res. 96, 6107-6112 (1991).
- 200
-
Ph. D'Arco, F. Freyria Fava, R. Dovesi and V.R. Saunders, ``Structural and
electronic properties of MgAlSiO pyrope garnet: an ab
initio study'',
J. Phys.: Cond. Matter 8, 8815-8828 (1996).
- 201
-
S. Dall'Olio, R. Dovesi, R. Resta, ``Spontaneous polarization as a Berry phase
of the Hartree-Fock wavefunction: The case of KNbO'',
Phys. Rev. B 56, 10105-10114 (1997).
- 202
-
F. Freyria Fava, Ph. D'Arco, R.Orlando and R.Dovesi, ``A quantum mechanical
investigation of the electronic and magnetic properties of CaMnO
perovskite'',
J. Phys.: Cond. Matter 9, 489-498 (1997).
- 203
-
C. Bluth and B. Paulus, ``Hydrogen bonding in infinite hydrogen fluoride and
hydrogen chloride chains'',
Phys. Rev. B 74, Art n. 045122 (2006).
- 204
-
Evarestov RA, Smirnov VP, Usvyat DE, ``Chemical bonding in crystalline silver
halides: Wannier-type atomic functions approach'',
Int. J. Quantum Chem. 96, 95-105 (2004).
- 205
-
M. Catti, A. Pavese, R. Dovesi, C. Roetti and M. Causà, ``Quantum-mechanical
Hartree-Fock-SCF study of the elastic constants and chemical bonding of
MgF (sellaite)'',
Phys. Rev. B 44, 3509-3517 (1991).
- 206
-
P. Azavant, A. Lichanot, M. Rerat and C. Pisani, ``A quantum mechanical
calculation of the dynamic structure factors of magnesium difluoride'',
Int. J. Quantum. Chem. 58, 419-429 (1995).
- 207
-
M. Catti, R. Dovesi, A. Pavese and V. R. Saunders, ``Elastic constants and
electronic structure of fluorite (CaF): an ab initio Hartree-Fock
study'',
J. Phys.: Condens. Matter 3, 4151-4164 (1991).
- 208
-
Puchina AV, Puchin VE, Huisinga M, Bennewitz R, Reichling M, ``Theoretical
modelling of steps and surface oxidation on CaF(111)'',
Surface Sci. 404, 687-691 (1998).
- 209
-
V.E. Puchin,A.V. Puchina, M. Huisinga, M .Reichling, ``Theoretical modeling of
steps on the CaF (111) surface'',
J. Phys. Condensed Matter 13, 2081-2094 (2001).
- 210
-
M. Huisinga, V.E. Puchin, M. Reichling, ``Photoemission from pure and electron
irradiated CaF'',
Nuc. Instr. Meth. B 141, 528-532 (1998).
- 211
-
Puchina AV, Puchin VE, Kotomin EA, Reichling M, ``Ab initio study of the F
centers in CaF: Calculations of the optical absorption, diffusion and
binding energies'',
Solid State Comm. 106, 285-288 (1998).
- 212
-
V.M. Bermudez, ``Ab-initio Hartree-Fock study of Mg
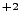 as a substitutional
impurity in CaF'',
as a substitutional
impurity in CaF'',
Solid State Comm. 118, 569-574 (2001).
- 213
-
Merawa M, Noel Y, Civalleri B, Brown R, Dovesi R, ``Raman and infrared
vibrational frequencies and elastic properties of solid BaFCl calculated with
various Hamiltonians: an ab initio study'',
J. Phys-Condems. Mat. 17, 535-548 (2005).
- 214
-
Bermudez V.M., ``Hartree-Fock study of near-edge gap states in CaF with
Na
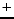 , Cl
, Cl or Sr
or Sr impurities'',
impurities'',
Comp. Mater. Sci 24, 501-512 (2002).
- 215
-
G. Valerio, M. Catti, R. Dovesi and R. Orlando, ``Ab initio study of
antiferromagnetic rutile-type FeF'',
Phys. Rev. B 52, 2422-2427 (1995).
- 216
-
I. de P.R. Moreira, R. Dovesi, C. Roetti, V.R. Saunders, R. Orlando, ``Ab
initio study of MF (M=Mn, Fe, Co, Ni) rutile-type compounds using the
periodic UHF approach'',
Phys. Rev. B 62, 7816-7823 (2000).
- 217
-
P. Reinhardt, M.P. Habas, R. Dovesi, I. de P.R. Moreira and F. Illas, ``On the
magnetic coupling in the weak ferromagnet CuF'',
Phys.Rev B 59, 1016-1023 (1999).
- 218
-
P. Reinhardt, I. de!P.R. Moreira, R. Dovesi, C. de Graaf and F. Illas,
``Detailed ab-initio analysis of the magnetic coupling in CuF'',
Chem. Phys. Lett. 319, 625-630 (2000).
- 219
-
M. Nizam, Y. Bouteiller, B. Silvi, C. Pisani, M. Causà and R. Dovesi, ``A
theoretical investigation of electronic structure and some thermodynamical
properties of -PbF'',
J. Phys. C: Solid State Phys. 21, 5351-5359 (1988).
- 220
-
R. Dovesi, F. Freyria Fava, C. Roetti and V.R. Saunders, ``Structural,
electronic and magnetic properties of KMF (M = Mn, Fe, Co, Ni)'',
Faraday Discuss. 106, 173-187 (1997).
- 221
-
J.M. Ricart, R. Dovesi, C. Roetti and V.R. Saunders, ``The electronic and
magnetic structure of KNiF perovskite'',
Phys. Rev. B 52, 2381-2389 (1995 (erratum ibid 55, 15942 (1997)).
- 222
-
M.D. Towler, R. Dovesi and V. R. Saunders, ``Magnetic interactions and the
co-operative Jahn-Teller effect in KCuF'',
Phys. Rev. B 52, 10150-10159 (1995).
- 223
-
R. Dovesi, J.M. Ricart, V.R. Saunders and R. Orlando, ``Superexchange
interaction in KNiF. An ab initio Hartree-Fock study'',
J. Phys.: Cond. Matter 7, 7997-8007 (1995).
- 224
-
M. Springborg, T. Asthalter and W. Weyrich, ``Electronic properties of the two
rutiles TiO and VF: A comparative study'',
Ber. Bunsenges. Phys. Chem. Chem. Phys. 102, 1760-1771
(1998).
- 225
-
Y. Noel, M. Llunell, R. Orlando, Ph. D'Arco, R. Dovesi, ``Performance of
various hamiltonians in the study of piezoelectric properties of crystalline
compounds: the case of BeO and ZnO'',
Phys. Rev. B 66, 214107 (2002).
- 226
-
A. Wander, C. L. Bailey, S. Mukhopadhyay, B. G. Searle and N. M. Harrison, ``Ab
initio studies of aluminium fluoride surfaces'',
J. Mater.Chem. 16, 1906-1910 (2006).
- 227
-
I. de P.R. Moreira, R. Dovesi, ``Ab initio periodic approach to electronic
structure and magnetic exchange in ACuOX (A=Ca, Sr and X=F,
Cl) High-T superconductor parent compounds'',
Phys. Rev. B 67, 134513 (2003).
- 228
-
Arrais A, Boccaleri E, Croce G, Milanesio M, Orlando R, Diana E, ``Synthesis,
structural and spectroscopic study of the donor-acceptor complexes between
fluorene and D-2h cyano molecular building blocks'',
CRYSTENGCOMM 5, 388-394 (2003).
- 229
-
J. Scaranto, G. Mallia, S. Giorgianni, C.M. Zicovich-Wilson, B. Civalleri, N.M.
Harrison, ``A quantum-mechanical study of the Vinyl Fluoride adsorbed on the
rutile TiO (110) surface'',
Surface Sci. 600, 305-317 (2006).
- 230
-
G. Croce, A. Arrais, E. Diana, B. Civalleri, D. Viterbo, M. Milanesio, ``The
interpretation of the short range disorder in the Fluorene-TCNE crystal
structure'',
Int. J. Mol. Sci. 5, 93-100 (2004).
- 231
-
P. Labéguerie, F. Pascale, M. Merawa, C.M. Zicovich Wilson, N. Makhoukhi, R.
Dovesi, ``Phononvibrational frequencies and elastic properties of solid
SrFCl. An ab initio study'',
European Physical J 43, 453-461 (2005).
- 232
-
Erikson RL, Early LE, Hostetlet CJ, ``ENERGETICS, STRUCTURE, AND
COMPRESSIBILITY OF NAF DETERMINED BY THE PERIODIC HARTREE-FOCK METHOD'',
J. Chem. Phys. 99, 336-344 (1993).
- 233
-
E. Aprà, M. Causà, M. Prencipe, R. Dovesi and V.R. Saunders, ``On the
structural properties of NaCl. An ab initio study of the B1-B2 phase
transition'',
J. Phys: Condens. Matter 5, 2969-2976 (1993).
- 234
-
Allouche A, ``Quantum ab initio study of acetylene adsorption on NaCl(100). 1.
Topology and adsorption energy'',
Surf. Sci 374, 117-124 (1997).
- 235
-
E.A. Mikajlo, K.L. Nixon, M.J. Ford, ``Electron momentum spectroscopy and
linear combination of atomic orbitals calculation of bulk NaO'',
J. Phys. Condens. Matter 15, 2155-2168 (2003).
- 236
-
E.A. Moore, C. Johnson, M. Mortimer,C. Wigglesworth, ``A comparison of ab
initio cluster and periodic calculations of the electric field gradient at
sodium in NaNO'',
Phys. Chem. Chem. Phys. 2, 1325-1331 (2000).
- 237
-
J.F.Sanz, C.M. Zicovich-Wilson, ``A periodic Hartree-Fock study of Na
adsorption on the TiO (110) rutile surface'',
Chem. Phys. Letters 303, 111-116 (1999).
- 238
-
A. Allouche, ``Water adsorption on NaCl(100): a quantum ab-initio cluster
calculation'',
Surf. Sci. 406, 279-293 (1998).
- 239
-
D.P. Taylor, W.P. Hess, M.I. McCarthy, ``Structure and energetics of the
water/NaCl(100) interface'',
J. Phys. Chem. B 101, 7455-7463 (1997).
- 240
-
C. Johnson, E.A. Moore, M. Mortimer, ``An assignement of the
 Na MAS NMR
spectrum of NaPO . 6HO using a general ab initio
method'',
Na MAS NMR
spectrum of NaPO . 6HO using a general ab initio
method'',
Chem. Commun., 791-792 (2000).
- 241
-
Florent Réal, Valérie Vallet, Jean-Pierre Flament, and Joël Schamps, ``Ab
initio study of a Bi
 impurity in CsNaYCl and YO:
Comparison of perturbative and variational electron correlation methods'',
impurity in CsNaYCl and YO:
Comparison of perturbative and variational electron correlation methods'',
J. Chem. Phys. 125, art n. 174709 (2006).
- 242
-
I. Baraille, C. Pouchan, M. Causà, F. Marinelli, ``Comparison between
Hartree-Fock and Kohn-Sham eletronic and structural properties for
exagonal-close-packed magnesium'',
J. Phys. Cond. Matter 10, 10969-10977 (1998).
- 243
-
M.P. Habas, R. Dovesi and A. Lichanot, ``B1-B2 phase transition in
alkaline-earth oxides: a comparison of ab initio Hartree-Fock and density
functional calculations'',
J. Phys. Cond. Matter 10, 6897-6909 (1998).
- 244
-
R. A. Evarestov, V. P. Smirnov, ``Symmetry of the model of a crystal with
periodic defect: point defects in MgO crystal'',
Phys. Stat. Sol. (b) 201, 75-87 (1997).
- 245
-
N. Cruz Hernandez, C. M. Zicovich-Wilson, Javier Fdez. Sanz, ``The constrained
space orbital variation analysis for periodic ab initio calculations'',
J. Chem. Phys. 124, 194105 (2006).
- 246
-
C. Pisani, F. Corà, R. Dovesi and R. Orlando, ``Embedded-cluster and
super-cell treatment of charged defects in crystals'',
J. Elec. Spec. 96, 1-12 (1994).
- 247
-
R. A. Evarestov, P. A. M. Jacobs, ``Oxygen interstitials in magnesium oxide: a
band-model study'',
Phys. Rev. B 54, 8969-8972 (1996).
- 248
-
L. Ojamae and C. Pisani, ``Theoretical characterization of divacancies at the
surface and in MgO bulk'',
J. Chem. Phys. 109, 10984-10995 (1998).
- 249
-
A. Lichanot, R. Orlando, G. Mallia, M. Merawa and R. Dovesi, ``V
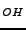 center
in magnesium oxide: an ab initio supercell study'',
center
in magnesium oxide: an ab initio supercell study'',
Chem. Phys. Lett. 318, 240-246 (2000).
- 250
-
C. Pisani, Ab-initio approaches to the quantum-mechanical treatment of
periodic systems, in Quantum-Mechanical Ab-initio Calculation of the
Properties of Crystalline Materials, edited by C. Pisani, Lecture
Notes in Chemistry, volume 67, 47-76, Springer Verlag, Berlin (1996).
- 251
-
M. Alfredsson, K. Hermansson, ``OH frequency calculations for the hydroxylated
MgO(001) surface'',
Molecular Simulation 28, 663-681 (2002).
- 252
-
Zhukovskii YF, Kotomin EA, Jacobs PWM, Stoneham AM, Harding JH, ``Modelling of
silver adhesion on MgO(100) surface with defects'',
J.Physics-Condensed Matter 12, 55-66 (2000).
- 253
-
Zhukovskii YF, Kotomin EA, Jacobs PWM, Stoneham AM, ``Ab initio modeling of
metal adhesion on oxide surfaces with defects'',
Phys. Rev. Letters 84, 1256-1259 (2000).
- 254
-
D. Fuks, S. Dorfman, Y.F. Zhukovskii, E.A. Kotomin, A.M. Stoneham, ``Theory of
the growth mode for a thin metallic film on an insulating substrate'',
Surface Sci. 499, 24-40 (2002).
- 255
-
Y.F. Zhukovskii, E.A. Kotomin, D. Fuks, S. Dorfman, A. Gordon, ``Hartree-Fock
study of adhesion and charge redistribution on the Ag/MgO(001) interface'',
Surface Sci. 482, 66-72 (2001).
- 256
-
B. Herschend, K. Hermansson, M. Alfredsson, Yu.F. Zhukovskii, E.A. Kotomin,
P.W.A. Jacobs, ``Characterization of the metal-ceramic bonding in the
Ag/MgO(001) interface from ab initio calculations'',
J. Phys. Chem. B 107, 11893-11899 (2003).
- 257
-
Yu.F. Zhukovskii, E.A. Kotomin, G. Borstel, ``Adsorption of single Ag and Cu
atoms on regular and defective MgO (001) substrates: an ab initio study'',
Vacuum 74, 235-240 (2004).
- 258
-
Zhukovskii YF, Kotomin EA, Jacobs PWM, Stoneham AM, Harding JH, ``Comparative
theoretical study of the Ag-MgO (100) and (110) interfaces'',
Surface Sci. 441, 373-383 (1999).
- 259
-
E.A. Kotomin, Yu.F. Zhukovskii, ``Ab initio modelling of metal adhesion to
ceramics with surface defects'',
Defects and Diffusion Forum 218-220, 67-78 (2003).
- 260
-
Yi Li, Yongfan Zhang, Liming Wu, Yijun Xu, Wenkai Chen and Junqian Li, ``A
theoretical study on the dissociation of Cl2 on MgO(0 0 1) surface: Prompted
by silver atoms supported on surface'',
Chem. Phys. 328, 236-242 (2006).
- 261
-
B. Herschend, M. Baudin, K. Hermansson, ``A combined molecular dynamics+quantum
mechanics method for investigation of dynamic effects on local surfaces
structures'',
J. Chem. Phys. 120, 4939-4948 (2004).
- 262
-
A.M. Ferrari, R. Soave, A. D'Ercole, C. Pisani, E. Giamello, G. Pacchioni,
``Theoretical characterization of charge-transfer reactions between N and
O molecules and paramagnetic oxygen vacancies on the MgO surface'',
Surface Sci. 479, 83-97 (2001).
gaussian.
- 263
-
A. Wander, N.M. Harrison, ``Stability of rocksalt polar surfaces: An ab initio
study of MgO(111) and NiO(111)'',
Phys. Rev. B 68, art n. 233405 (2003).
- 264
-
E. Scorza, U. Birkenheuer and C. Pisani, ``The oxygen vacancy at the surface
and in bulk MgO: an embedded-cluster study'',
J. Chem. Phys. 107, 9645-9659 (1997).
- 265
-
G. Pinarello, C. Pisani, A. D'Ercole, M. Chiesa, M.C. Paganini, E. Giamello,
O. Diwald, ``O radical ions on MgO as a tool to unravel structure and
location of ionic vacancies at the surface of oxides. A coupled experimental
and theoretical investigation.'',
Surface Sci. 491, 95-110 (2001).
- 266
-
F. Illas, G. Pacchioni, A. Pelmenschikov, L.G.M. Pettersson, R. Dovesi, C.
Pisani, K.M. Neyman, N. Rösch, ``Comment on "First principles
determination of the bonding mechanism and adsorption energy for
CO/MgO(001)"'',
Chem. Phys. Letters 206, 202-204 (1999).
- 267
-
A. Damin, R. Dovesi, A. Zecchina, P. Ugliengo, ``CO/MgO(001) at different CO
coverages: a periodic ab initio Hartree-Fock and B3-LYP study'',
Surface Sci. 479, 253-272 (2001).
- 268
-
P. Ugliengo, A. Damin, ``Are dispersive forces relevant for CO adsorption on
the MgO (001) surface?'',
Chem. Phys. Lett. 366, 683-690 (2002).
- 269
-
Chacon-Taylor MR, McCarthy MI, ``Ab initio based classical electrostatic
potentials for the interaction between molecules and surfaces'',
J. Phys. Chem. 100, 7610-7616 (1996).
- 270
-
A. Shluger, C. Pisani, C. Roetti and R. Orlando, ``Ab initio simulation of the
interaction between ionic crystal surfaces and the atomic force microscope
tip'',
J. Vac. Sci. Technol. A8, 3967-3972 (1990).
- 271
-
E. Kotomin, A. Shluger, M. Caus`, R. Dovesi and F. Ricca, ``The adsorption of
SiO molecules on MgO surfaces as a model for the silicon lever atomic force
microscope (AFM)'',
Surf. Sci. 232, 399-406 (1990).
- 272
-
Markovits A, Ahdjoudj J, Minot C, ``Theoretical study of the adsorption of
acids and bases on TiO and MgO surfaces'',
Nuovo Cimento D 19, 1719-1726 (1997).
- 273
-
Gracia L, Beltran A, Andres J, Franco R, Recio JM, ``Quantum-mechanical
simulation of MgAlO under high pressure'',
Phys. Rev. B 66, art no 224114 (2002).
- 274
-
Beltran A, Gracia L, Andres J, Franco R, Recio JM, ``Stability of MgAlO
under high-pressure conditions'',
High Pressure Res 22, 447-450 (2002).
- 275
-
L. Benco, L. Smrcok, ``Hartree-Fock study of pressure-induced strengthening of
hydrogen bonding in lizardite-1T'',
European Journal of Mineralogy 10, 483-490 (1998).
- 276
-
N.M. Harrison and V.R. Saunders, ``The structural properties of
-MgCl: an ab initio study.'',
J. Phys. Cond. Matter 4, 3873-3882 (1992).
- 277
-
N.M. Harrison, V.R. Saunders, E. Aprà, M. Causà and R. Dovesi,
``Correlation functional estimates of the dispersion interaction in
semi-ionic compounds'',
J. Phys.: Condens. Matter 4, L261-L264 (1992).
- 278
-
Barrera GD, Allan NL, Soriano MR, ``The stability of polymorphs of MgCl'',
Chem. Phys. Letters 278, 267-271 (1997).
- 279
-
Harrison NM, Leslie M, ``The derivation of shell-model potentials for MgCl
from ab initio theory'',
Molecular Simulation, 171-174 (1992).
- 280
-
T.N.P. Luhtanen, M. Linnolahti, T.A. Pakkanen, ``Molecular structures of
magnesium dichloride sheets and nanoballs'',
Inorg. Chem 43, 4482-4486 (2004).
- 281
-
Linnolahti M, Pakkanen TA, ``Quantum chemical treatment of large nanotubes via
use of line group symmetry: Structural preferences of magnesium dichloride
nanotubes'',
J. Phys. Chem. B 110, 4675-4678 (2006).
- 282
-
Soriano MR, Barrera GD, Allan NL, ``Vibrational energies and thermal expansion
of layered compounds: MgCl'',
Chem. Phys. Letters 350, 543-550 (2001).
- 283
-
M. Konigstein F. Corà and C.R.A. Catlow, ``An ab initio Hartree-Fock study of
the energies of mixing of MnO-NiO, MgO-MnO, and CaO-MnO solid solutions'',
J. Solid State Chem 137, 261-275 (1998).
- 284
-
Ferrari AM, Casassa S, Pisani C, Altieri S, Rota A, Valeri S, ``Polar and
non-polar domain borders in MgO ultrathin films on Ag(001)'',
Surface Sci. 588, 160-166 (2005).
- 285
-
Ph. Baranek, A. Lichanot, R. Orlando, R. Dovesi, ``Structural and vibrational
properties of solid Mg(OH) and Ca(OH) - Performances of various
hamiltonians'',
Chem. Phys. Letters 240, 362-369 (2001).
- 286
-
A.G. Chizmeshya, M.J. McKelvy, R. Sharma, R.W. Carpenter, i H. Bearat,
``Density functional theory study of the decomposition of Mg(OH): a
lamellar dehydroxylation model'',
Materials Chemistry and Physics 77, 416-425 (2003).
- 287
-
Barrera GD, Taylor MB, Allan NL, Barron THK, Kantorovich LN, Mackrodt WC,
``Ionic solids at elevated temperatures and high pressures: MgF'',
J. Chem. Phys. 107, 4337-4344 (1997).
- 288
-
Allan NL, Hines RI, Towler MD, Mackrodt WC, ``Calculated pressure-induced phase
transition in MgF'',
J. Chem. Phys. 100, 4710-4711 (1994).
- 289
-
A. Lichanot, A. Dargelos, C. Larrieu and R. Orlando, ``Electronic structure and
phase transition in magnesium sulphide'',
Solid State Comm. 90, 189-193 (1994).
- 290
-
M. Catti, A. Pavese, ``Theoretical structure factors and electron density of
magnesite MgCO'',
Acta Crystallogr. A52, 413-418 (1996).
- 291
-
M. Schmidth, W. Ratcliff, P.G. Radaelli, K. Refson, N.M. Harrison, S.W. Cheong,
``Spin singlet formation in MgTiO: evidence of helical dimerization
pattern'',
Phys. Rev. Letters 92, art n. 056402 (2004).
- 292
-
G. Ottonello, B. Civalleri, M. Vetuschi Zuccolini, C.M. Zicovich-Wilson,
``Ab-initio thermal physics and Cr-isotopic fractionation of MgCRO'',
American Mineralogist 92, 98-108 (2007).
- 293
-
M. Catti, ``High-pressure stability, structure and compressibility of
Cmcm-MgAlO: an ab initio study'',
Phys. Chem. Miner. 28, 729-736 (2001).
- 294
-
P. Baranek and J. Schamps, ``Influence of electronic correlation on structural,
dynamic and elastic properties of MgSi'',
J. Phys. Chem. B 103, 2601-2606 (1999).
- 295
-
S. Casassa and C. Pisani, ``Interaction of HOCl with a chlorinated ice surface
to produce molecular chlorine: an ab-initio study.'',
J. Chem. Phys. 116, 9856-9864 (2002).
- 296
-
Y. R. Zhukovskii, E.A. Kotomin, D. Fuks, S. Dorfman, M. Stoneham, O. Sychev,
G. Borstel, ``First Principles simulations of 2D Cu superlattices on the
MgO(001) surface.'',
Applied Surf. Sci. 226, 298-305 (2004).
- 297
-
Maslyuk V, Tegenkamp C, Pfnur H, Bredow T, ``Adsorption of functionalized
benzoic acids on MgSO center dot HO100'',
ChemPhysChem 7, 1055-1061 (2006).
- 298
-
F.J. Torres, B. Civalleri, A. Terentyev, P. Ugliengo, C. Pisani, ``Theoretical
study of molecular Hydrogen adsorption in Mg-Exchanged chabazite'',
J. Phys. Chem. C 111, 1871-1873 (2007).
- 299
-
Bredow T, Gerson AR, ``Effect of exchange and correlation on bulk properties of
MgO, NiO, and CoO'',
Phys. Rev. B 61, 5194-5201 (2000).
- 300
-
A.M. Ferrari, C. Roetti, C. Pisani, ``Water dissociation at MgO sub-monolayers
on silver: a periodic model study'',
Phys. Chem. Chem. Phys. 9, 2350-2354 (2007).
- 301
-
F Zhukovskii, Eugene A Kotomin, David Fuks, Simon Dorfman, A Marshall Stoneham
and Gunnar Borstel, ``Adhesion trends and growth mode of ultra-thin copper
films on MgO'',
J. Phys.: Condens. Matter 16, 4881-4896 (2004).
- 302
-
F Zhukovskii, Eugene A Kotomin, David Fuks, Simon Dorfman, ``First principles
slab calculations of the regular Cu/MgO(001) interface'',
Surface Sci. 566-568, 122-129 (2004).
- 303
-
Smirnov VP, Evarestov RA, ``Symmetry analysis for localized function generation
and chemical bonding in crystals: SrZrO and MgO as examples'',
Phys. Rev. B 72, Art. N. 075138 (2005).
- 304
-
Evarestov RA, Smirnov VP, Usvyat DE, ``Wannier functions and chemical bonding
in a slab model: MgO(001) and TiO(110) surfaces'',
Int. J. Quantum Chem. 104, 102-109 (2005).
- 305
-
M. Causà, R. Dovesi, C. Pisani and C. Roetti, ``Exact exchange Hartree-Fock
calculations for periodic systems. Directional Compton profiles of
aluminum'',
Philos. Mag. 44, 419-425 (1981).
- 306
-
B. Montanari, B. Civalleri, C.M. Zicovich-Wilson, R. Dovesi, ``Influence of the
exchange-correlation functional in all-electron calculations of the
vibrational frequencies of Corundum (-AlO)'',
Int. J. Quantum Chem. 106, 1703-1714 (2006).
- 307
-
C. Rinaudo, P. Orione, M. Causà, ``Verneuil corundum: an integrated PBC and
white beam synchrotron radiation X-ray topography analysis'',
J. Crystal Growth 244, 53-62 (2002).
- 308
-
F. Janetzko, R.A. Evarestov, T. Bredow, K. Jug, ``First-principles periodic and
semiempirical cyclic cluster calculations for single oxygen vacancies in
crystalline
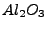 '',
'',
Phys. Stat. Sol. (b) 241, 1032-1040 (2004).
- 309
-
Puchin VE, Gale JD, Shluger AL, Kotomin EA, Gunster J, Brause M, Kempter V,
``Atomic and electronic structure of the corundum(0001) surface: Comparison
with surface spectroscopies'',
Surface Sci. 370, 190-200 (1997).
- 310
-
Puchin VE, Gale JD, Shluger AL, Kotomin EA, Gunster J, Brause M, Kempter V,
``Atomic structure of the (0001) corundum surface'',
Mater Sci. Forum 239, 633-636 (1997).
- 311
-
J.R.B. Gomes, I.de P.R. Moreira, P. Reinhardt, A. Wander, B.G. Searle,
N.M. Harrison, F. Illas, ``The structural relaxation of the
-AlO (0001) - an investigation of potential errors'',
Chem. Phys. Lett. 341, 412-418 (2001).
- 312
-
Y. F. Zhukovskii, E.A. Kotomin, B. Herschend, K. Hermansson, P.W.M. Jacobs,
``The adhesion properties of the Ag/-AlO(0 0 0 1) interface:
an ab initio study'',
Surface Sci 24 May, - (2002).
- 313
-
A. Wander, B. Searle, N.M. Harrison, ``An ab initio study of
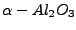 (0001): the effects of exchange and correlation functionals.'',
(0001): the effects of exchange and correlation functionals.'',
Surface Sci. 458, 25-33 (2000).
- 314
-
Nagy LT, Micov M, Benco L, Liska M, Mach P, Tunega D, ``Electronic structure of
alumina surface'',
Int. J. Quantum Chem. 70, 341-350 (1998).
- 315
-
Baxter R, Reinhardt P, Lopez N, Illas F, ``The extent of relaxation of the
-AlO (0001) surface and the reliability of empirical
potentials'',
Surface Sci. 445, 448-460 (2000).
- 316
-
Hess AC, Saunders VR, ``Periodic ab initio Hartree-Fock calculations of the low
symmetry mineral kaolinite'',
J. Phys. Chem. 96, 4367-4374 (1992).
- 317
-
A. Hung, S.P. Russo, D.G. McCulloch, ``An ab-initio study of structural
properties and single vacancy defects in Wurtzite AlN'',
J. Chem. Phys. 120, 4890-4896 (2004).
- 318
-
Yu.F. Zhukovskii, P.W.M. Jacobs, M. Causá, ``On the mechanism of the
interaction between oxygen and close-packed single-crystal aluminum
surfaces'',
J. Phys. Chem. Solids 64, 1317-1331 (2003).
- 319
-
F. Pascale, P. Ugliengo, B. Civalleri, R. Orlando, P. D'Arco, R. Dovesi, ``The
katoite hydrogarnet Si-free CaAl([OH]): a periodic
Hartree-Fock and B3-LYP study'',
J. Chem. Phys. 121, 1005-1013 (2004).
- 320
-
R. Orlando, F.J. Torres, F. Pascale, P. Ugliengo, C.M. Zicovich-Wilson, R.
Dovesi, ``Vibrational spectrum of katoite CaAl[(OH)]: a
periodic ab initio study'',
J. Phys. Chem. B 110, 692-701 (2006).
- 321
-
Saadoune I, Catlow CRA, Doll K, Cora F, ``Water adsorption and acidity in
Mn-II-HAlPO-34 catalysts'',
Mol. Simulat. 30, 607-615 (2004).
- 322
-
F. Corà, I. Saadoune, C.R.A. Catlow, ``Lewis acidity in
transition-metal-doped microporous aluminophosphates'',
Angew. Chem. Int. Edit. 41, 4677-4680 (2002).
- 323
-
Yu.F. Zhukovskii, E.A. Kotomin, Yu. Mastrikov and J. Maier, ``Ab initio
simulations on AgCl(1 1 1) surface and AgCl(1 1 1)/?-Al2O3(0 0 0 1)
interface'',
Comp. Mat. Science 33, 276-281 (2005).
- 324
-
R. Dovesi, M. Causà and G. Angonoa, ``Exact exchange Hartree-Fock
calculations for periodic systems. V. Ground state properties of silicon'',
Phys. Rev. B 24, 4177-4183 (1981).
- 325
-
G. Angonoa, R. Dovesi, C. Pisani and C. Roetti, ``Exact exchange Hartree-Fock
calculations for periodic systems. Directional Compton profiles of silicon in
the impulse approximation'',
Philos. Mag. 44, 413-418 (1981).
- 326
-
Markovits A, Favaro L, Minot C, ``Hartree-Fock study of the Si(100)
reconstruction'',
Theochem-J. Molec. Structur. 458, 171-189 (1999).
- 327
-
R. Lindsay, P.L. Wincott, G. Thornton, N.M. Harrison, ``The electronic
structure of Si(100)2x1-Cl: reinterpreting ARP measurements'',
Surf. Sci. 398, 301-307 (1998).
- 328
-
C. Pisani, R. Dovesi and R. Orlando, ``Near-Hartree-Fock wave functions for
solids: the case of crystalline silicon'',
Int. J. Quantum Chem. 42, 5-33 (1991).
- 329
-
Sonnet P; Stauffer L; Minot C, ``Periodic Hartree-Fock calculation of the
oxidation of Si(111)'',
Surface Review and Letters 6, 1031-1036 (1999).
- 330
-
Tse JS, Dahn JR, Buda F, ``Electronic structure of layer polysilanes'',
J. Phys. Chem 89, 1896-1899 (1995).
- 331
-
D. Ayma, M. Rerat, R. Orlando, A. Lichanot, ``Ab initio calculation of the
structure factors and Compton profiles of cubic silicon carbid'',
Acta Cryst. A 54, 1019-1027 (1998).
- 332
-
A.B. Mukhopadhyay, M. Dolg, C. Oligschleger, ``Ab initio many-body
investigation of structure and stability of two-fold rings in silicates.'',
J. Chem. Phys. 120, 8734-8739 (2004).
- 333
-
M. Busso, S. Casassa, C. Pisani and V.B. Sulimov, ``Ab-initio simulation of
the oxygen vacancy instability in pure and Ge-doped quartz.'',
Modelling Simul. Mater. Sci. Eng. 10, 21-33 (2002).
- 334
-
Gibbs GV; Rosso KM; Teter DM; Boisen MB; Bukowinski MST, ``Model structures and
properties of the electron density distribution for low quartz at pressure: a
study of the SiO bond'',
J. Mol. Struct. 486, 13-25 (1999).
- 335
-
B. Civalleri, C.M. Zicovich-Wilson, P. Ugliengo, V.R. Saunders and R. Dovesi,
``A periodic ab-initio study of the structure and relative stability of
silica polymorphs'',
Chem. Phys. Lett 292, 394-402 (1998).
- 336
-
M. Catti, B. Civalleri, P. Ugliengo, ``Structure and energetics of
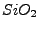 polymorphs by quantum-mechanical and semiclassical approaches'',
polymorphs by quantum-mechanical and semiclassical approaches'',
J. Phys. Chem B 104, 7259-7265 (2000).
- 337
-
M. A. Zwijnenburg, F. Corà, R.G. Bell, ``On the performance of DFT and
interatomic potentials in predicting the energetics of (three-membered
ring-containing) silicious materials'',
J. Phys. Chem. B 111, 6156-6160 (2007).
- 338
-
B. Civalleri, S. Casassa, E. Garrone, C. Pisani and P. Ugliengo, ``Quantum
mechanical ab-initio characterization of a simple periodic model of the
silica'',
J. Phys. Chem. 103, 2165-2171 (1999).
- 339
-
G.V. Gibbs, K.M. Rosso, D.M. Teter, M.B. Boisen and M.S.T. Bukowinski, ``Model
structure and electron density distribution of the silica polymorph coesite
at pressure: An assessment of OO bonded interactions.'',
J. Phys. Chem. B 104, 10534-10542 (2000).
- 340
-
G.V. Gibbs, A.E. Whitten, M.A. Spackam, M. Stimpfl, R.T. Downs, M.D. Carducci,
``An exploration of theoretical and esperimental electron density
distributions and SiO bonded interactions for the silica polymorph coesite'',
J. Phys. Chem. B 107, 12996-13006 (2003).
- 341
-
G. V. Gibbs, M. A. Spackman, D. Jayatilaka, K. M. Rosso, and D. F. Cox, ``Bond
Length and Local Energy Density Property Connections for Non-Transition-Metal
Oxide-Bonded Interactions'',
J. Phys. Chem. A 110, 12259-12266 (2006).
- 342
-
Gnani E; Reggiani S; Colle R; Rudan M, ``Band-structure calculations of SiO2 by
means of Hartree-Fock and density-functional techniques'',
IEEE TRANSACTIONS ON ELECTRON DEVICES 47, 1795-1803
(2000).
- 343
-
V. Sulimov, S. Casassa, C. Pisani, J. Garapon, B. Poumellec, ``Embedded cluster
ab initio study of the neutral oxygen vacancy in quartz and cristobalite'',
Modelling Simul. Mater. Sci. Eng. 8, 763-773 (2000).
- 344
-
L. Benco, L. Smrcok, ``Ab initio calculated electron densities for tetrahedral
sheet [HSiO]
 '',
'',
Phys. Chem. Minerals 21, 401-406 (1994).
- 345
-
K.M. Rosso, G.V. Gibbs, M.B. Boisen, ``SiO bonded interactions in coesite: a
comparison of crystalline, molecular and experimental electron density
distributions'',
Physics and Chemistry of minerals 26, 264-272 (1999).
- 346
-
G.V. Gibbs, K.M. Rosso, D.M. Teter, M.B. Boisen and M.S.T. Bukowinski, ``Model
structures and properties of the electron density distribution of low quartz
at pressure: A study of the SiO bond'',
J. Mol. Struct. 485-486, 13-25 (1999).
- 347
-
I. Baraille, M. Loudet, S. Lacombe, H. Cardy, C. Pisani, ``Chemisorption of
HSi(OH) on silica surfaces: an ab initio periodic study'',
J. Molec. Struc. THEOCHEM 620, 291-300 (2003).
- 348
-
F. Lopez-Gejo, M. Busso, C. Pisani, ``Quantum mechanical ab initio study of
mixed SiO-GeO crystal as reference models for Ge-doped silica
glasses'',
J. Phys. Chem. B 107, 2944-2952 (2003).
- 349
-
Zwijnenburg MA, Huenerbein R, Bell RG, Cora F, ``A computational study into the
(tetrahedral) distortion of TX alpha-quartz materials: The effect of
changing the chemical composition away from SiO'',
J. Solid State Chem. 179, 3429-3436 (2006).
- 350
-
D. Zur, D. Menzel, I. Jursic, J. Schoenes, L. Patthey, M. Neef, K. Doll, and G.
Zwicknagl, ``Absence of Kondo resonance in high-resolution photoemission
spectra of monocrystalline FeCoSi'',
Phys. Rev. B 75, Art. N. 165103 (2007).
- 351
-
T. P. M. Goumans, Adrian Wander, Wendy A. Brown and C. Richard A. Catlow,
``Structure and stability of the (001) -quartz surface'',
Phys. Chem. Chem. Phys. 9, 2146-2152 (2007).
- 352
-
Knaup JM, Deak P, Frauenheim T, Gali A, Hajnal Z, Choyke WJ, ``Defects in
SiO as the possible origin of near interface traps in the SiC/SiO
system: A systematic theoretical study'',
Phys. Rev. B 72, Art. N. 115323 (2005).
- 353
-
Knaup JM, Deak P, Gali A, Hajnal Z, Frauenheim T, Choyke JW, ``The search for
near interface oxide traps - First-principles calculations on intrinsic
SiO defects'',
SILICON CARBIDE AND RELATED MATERIALS 2004 483, 569-572
(2005).
- 354
-
C. Zicovich-Wilson and R. Dovesi, ``Periodic ab-initio study of the oxidizing
sites in Ti-containing Zeolites'',
J. Molec. Catal. A: Chemical 119, 449-458 (1997).
- 355
-
E. Aprà, R. Dovesi, C. Freyria-Fava, C. Pisani, C. Roetti and V.R. Saunders,
``Ab initio Hartree-Fock modelling of zeolites: application to
silico-chabazite'',
Modelling. Simul. Mater. Sci. Eng. 1, 297-306 (1993).
- 356
-
P. Ugliengo, B. Civalleri, C.M. Zicovich-Wilson and R. Dovesi, ``H-chabazite
with variable Si/Al ratio: stability and OH vibrational frequency computed in
a periodic LCAO B3-LYP approach'',
Chem. Phys. Lett. 318, 247-255 (2000).
- 357
-
E.H. Teunissen, C. Roetti, A.J.M. De Man, A.P.J. Jansen, R. Orlando,
R.A. Van Santen and R. Dovesi, ``Proton transfer in zeolites: a comparison
between cluster and periodic calculations'',
Modelling. Simul. Mater. Sci. Eng. 2, 921-932 (1994).
- 358
-
C. Pisani and U. Birkenheuer, ``Embedded-cluster approach to the study of
catalytic reactions in zeolite cavities'',
Int. J. Quantum. Chem. QCS 29, 221-234 (1995).
- 359
-
C. Zicovich-Wilson, R. Dovesi, ``Periodic ab-initio study of
silico-faujasite'',
Chem. Phys. Lett. 277, 227-233 (1997).
- 360
-
E.H. Teunissen, A.P.J. Jansen, R.A. van Santen, R. Orlando and R. Dovesi,
``Adsorption energies of NH and NH
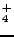 in zeolites corrected for the
long-range electrostatic potential of the crystal'',
in zeolites corrected for the
long-range electrostatic potential of the crystal'',
J. Chem. Phys. 101, 5865-5874 (1994).
- 361
-
M. Br
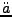 ndle, J. Sauer, R. Dovesi and N.M. Harrison, ``Comparison of a
combined quantum mechanics/interatomic potential function approach with its
periodic quantum mechanical limit. Proton siting and ammonia adsorption in
zeolite chabazite'',
ndle, J. Sauer, R. Dovesi and N.M. Harrison, ``Comparison of a
combined quantum mechanics/interatomic potential function approach with its
periodic quantum mechanical limit. Proton siting and ammonia adsorption in
zeolite chabazite'',
J. Chem. Phys. 109, 10379-10389 (1998).
- 362
-
C. Zicovich-Wilson and R. Dovesi, ``On the structure and stability of
Ti-Zeolites. A comparison of cluster and periodic ab initio
calculations.'',
Nuovo Cimento 19, 1785-1790 (1997).
- 363
-
C. Zicovich-Wilson and R. Dovesi, ``Titanium containing zeolites. A periodic
ab-initio Hartree Fock characterization'',
J. Phys. Chem. 102, 1411-1417 (1998).
- 364
-
C.M. Zicovich-Wilson, R. Dovesi and A. Corma, ``Interaction of Ti-Zeolites with
water. A periodic ab initio study'',
J. Phys. Chem. B 103, 988-994 (1999).
- 365
-
A. Damin, F.X. Llabres i Xamena, C. Lamberti, B. Civalleri,
C.M. Zicovich-Wilson,n A. Zecchina, ``Structural, electronic and vibrational
properties of the titanosilicate ETS-10: an ab-initio periodic study'',
J. Phys. Chem. B 108, 1328-1336 (2004).
- 366
-
P. Ugliengo, B. Civalleri, R. Dovesi and C.M. Zicovich-Wilson, ``Periodic
B3-LYP calculations of H-edingtonites both freee and interacting with
acetylene'',
Phys. Chem. Chem. Phys 1, 545-553 (1999).
- 367
-
J.C. White, J.B. Nicholas and A.C. Hess, ``Periodic Hartree-Fock
characterization of the structure and electronic properties of zeolite
NaCaA'',
J. Phys. Chem. B 101, 590-595 (1997).
- 368
-
F. Marquez, C.M. Zicovich-Wilson, A. Corma, E. Palomares, H. Garcia,
``Naphtalene included within all-silica zeolites: influence of the host on
the naphtalene photophysics'',
J. Phys. Chem. B 105, 9973-9979 (2001).
- 369
-
F. Pascale, P. Ugliengo, B. Civalleri, R. Orlando, Ph. D'Arco, R. Dovesi,
``Hydrogarnet defect in chabazite and sodalite zeolites: a periodic
Hartree-Fock and B3-LYP'',
J. Chem. Phys. submitted, 5337-5346 (2002).
- 370
-
A. Damin, S. Bordiga, A. Zecchina, K. Doll, C. Lamberti, ``Ti-chabazite as a
model system of Ti(IV) in Ti-zeolites: A periodic approach.'',
J. Chem. Phys. 118, 10183-10194 (2003).
- 371
-
F. Corà, C.R.A. Catlow, B. Civalleri, R. Orlando, ``Acid strength of
low-valence dopant ions in microporous zeolites and AlPOs'',
J. Phys. Chem. B 107, 11866-11870 (2003).
- 372
-
Cora F, Catlow CRA, ``Influence of the counterion on the local environment and
electronic structure of active sites in zeotypes'',
J. Phys. Chem. B 107, 11861-11865 (2003).
- 373
-
Cora F, Alfredsson M, Barker CM, Bell RG, Foster MD, Saadoune I, Simperler A,
Catlow CRA, ``Modeling the framework stability and catalytic activity of pure
and transition metal-doped zeotype'',
J. Solid State Chem. 176, 496-529 (2003).
- 374
-
B. Civalleri, P. Ugliengo, ``First principle calculations of the adsorption of
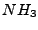 on a periodic model of silica surface'',
on a periodic model of silica surface'',
J. Phys. Chem. B 104, 9491-9499 (2000).
- 375
-
Hong SB, Lee SH, Shin CH, Woo AJ, Alvarez LJ, Zicovich-Wilson CM, Camblor MA,
``In situ disorder-order transformation in synthetic gallosilicate zeolites
with the NAT topology'',
J. Am. Chem. Soc. 126, 13742-13751 (2004).
- 376
-
X. Solans-Monfort, M. Sodupe, V. Branchadell, J. Sauer, R. Orlando, P.
Ugliengo, ``Adsorption of NH and HO in acidic chabazite. Comparison
of ONIOM approach with periodic calculations'',
J. Phys. Chem. 109, 3539-3545 (2005).
- 377
-
P. Ugliengo, C. Busco, B. Civalleri, C. M. Zicovich-Wilson, ``Carbon monoxide
adsorption on alkali and proton-exchanged chabazite: an ab-initio periodic
study using the CRYSTAL code'',
Mol. Phys. 103, 2559-2571 (2005).
- 378
-
V. Bolis, A. Barbaglia, M. Broyer, C. Busco, B. Civalleri, P. Ugliengo,
``Entrapping molecules in zeolites nanocavities: a thermodynamic and
ab-initio study'',
Origins of Life and Evolution of the Biosphere 34,
69-77 (2004).
- 379
-
A. Lichanot and M. Causà, ``Compared electronic charge densities for the
series of solids phosphide compounds: an ab-initio study'',
J. Phys. Cond. Matter 9, 3139-3149 (1997).
- 380
-
A. Pedone, M. Corno, B. Civalleri, G. Malavasi, M. C Menziani, U. Segre and P.
Ugliengo, ``An ab initio parameterized interatomic force field for
hydroxyapatite'',
J. Mat. Chem. 17, 2061-2068 (2007).
- 381
-
R. Tappero, I. Baraille, A. Lichanot, ``Electronic structure of pyrite-type
manganese disulphide (pMnS(2)): An ab initio study'',
Phys. Rev B 58, 1236-1242 (1998).
- 382
-
Tappero R; Wolfers P; Lichanot A, ``Electronic, magnetic structures and neutron
diffraction in B-1 and B-3 phases of MnS: a density functional approach'',
Chem. Phys. Letters 335, 449-457 (2001).
- 383
-
A. Ibarz E. Ruiz, S. Alvarez, ``Electronic structure of host lattices for
intercalation compounds: SnS, SnSe, ZrS, and TaS'',
Chemistry of materials 10, 3422-3428 (1998).
- 384
-
D.G. Clerc, R.D. Poshusta and A.C. Hess, ``Periodic Hartree-Fock study of
TiS'',
J. Phys. Chem. 100, 15735-15747 (1996).
- 385
-
E. Amzallag, I. Baraille, H. Martine, M. Rérat, M. Loudet, D. Gonbeau,
``Electronic and structural properties of Ti vacancies on the (001) surface
of TiS: theoretical scanning microscopy images'',
J. Chem. Phys. 126, Art. N. 074703 (2007).
- 386
-
K.M. Rosso, M.F. Hochella, ``A UHV STM/STS and ab initio investigation of
covellite (001) surfaces'',
Surface Sci. 423, 364-374 (1999).
- 387
-
A. Masunov and J.J. Dannenberg, ``Theoretical study of urea and thiourea. 2.
Chains and ribbons.'',
J. Phys. Chem. B 104, 806-810 (2000).
- 388
-
M. Catti, Y. Noel, R. Dovesi, ``Full piezoelectric tensor of wurtzite and zinc
blende ZnO and ZnS by first-principles calculations'',
J. Phys. Chem. Solids 64, 2183-2190 (2003).
- 389
-
M. Catti, ``First-principles study of the orthorhombic mechanism for the B3/B1
high-pressure phase transition of ZnS'',
Phys. Rev. B 65, art 224115 (2002).
- 390
-
Mian M, Harrison NM, Saunders VR, Flavell WR, ``An ab initio Hartree-Fock
investigation of galena (PbS)'',
Chem. Phys. Letters 257, 627-632 (1996).
- 391
-
J. Muscat, C. Klauber, ``A combined ab initio and photoelectron study of galena
(PbS)'',
Surface Sci. 491, 226-238 (2001).
- 392
-
J. Muscat, J.D. Gale, ``First principles studies of the surface of galena
PbS'',
Geochim. Cosmochim. Ac. 67, 799-805 (2003).
- 393
-
U. Becker, K.M. Rosso, ``Step edges on galena (100): Probing the basis for
defect driven surface reactivity at the atomic scale'',
American Mineralogist 86, 862-870 (2001).
- 394
-
J. Muscat, A. Hung, S. Russo, I. Yarovsky, ``First-principle studies of the
structural and electronic properties of pyrite
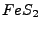 '',
'',
Phys. Rev. B 65, n. 054107 (2002).
- 395
-
Hung A, Muscat J, Yarovsky I, Russo SP, ``Density-functional theory studies of
pyrite FeS2 (111) and (210) surfaces'',
Surface Sci. 520, 111-119 (2002).
- 396
-
Hung A, Muscat J, Yarovsky I, Russo SP, ``Density-functional theory studies of
pyrite FeS2 (100) and (110) surfaces'',
Surface Sci. 513, 511-524 (2002).
- 397
-
K.M. Rosso, U. Becker, M.F. Hochella, ``Atomically resolved electronic
structure of pyrite (100) surfaces: An experimental and theoretical
investigation with implications for reactivity.'',
Am. Min. 84, 1535-1548 (1999).
- 398
-
K.M. Rosso, U. Becker, M.F. Hochella, ``The interaction of pyrite (100)
surfaces with O2 and H2O: fundamental oxidation mechanisms.'',
Am. Min. 84, 1549-yyy (1999).
- 399
-
G.V. Gibbs, D.F. Cox, K.M. Rosso, N.L. Ross, R.T. Downs, M.A. Spackman,
``Theoretical electron density distributions for Fe- and Cu- sulfide earth
materials: a connection between bond-length, bond critical point properties,
local energy densities, and bond interactions'',
J. Phys. Chem. B 111, 1923-1931 (2007).
- 400
-
R. Weihrich, I. Anusca, ``Halbantiperovskites II: on the structure of
PdBiS'',
Z. Anorg. Allg. Chem. 632, 335-342 (2006).
- 401
-
E. Aprà, E. Stefanovich, R. Dovesi and C. Roetti, ``An ab initio Hartree-Fock
study of silver chloride'',
Chem. Phys. Lett. 186, 329-335 (1991).
- 402
-
I. Noiret, F. Baert, G. Odou, F. Danede, J. Schamps and P. Baranek, ``Rubidium
tetrachlorocadmate: X-ray diffraction measurements and an electronic
structure study'',
J. Solid State Chem. 140, 371-376 (1998).
- 403
-
K. Doll and N.M. Harrison, ``Chlorine adsorption on the Cu(111) surface'',
Chem. Phys. Lett. 317, 282-289 (2000).
- 404
-
K. Doll, ``Density functional study of Ni bulk, surfaces and the adsorbate
systems Ni(111)(
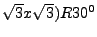 -Cl and Ni(111)(2 x 2) -K'',
-Cl and Ni(111)(2 x 2) -K'',
Surface Sci. 544, 103-120 (2003).
- 405
-
Marabello D, Bianchi R, Gervasio G, Cargnoni F, ``An experimental (120 K) and
theoretical electron-density study of KMnOand KClO'',
ACTA CRYSTALLOGRAPHICA A 60, 494-501 (2004).
- 406
-
T. Bredow, E. Aprà, M. Catti, G. Pacchioni, ``Cluster and periodic ab-initio
calculations on K/TiO(110)'',
Surf. Sci. 418, 150-165 (1998).
- 407
-
Lindsay R, Michelangeli E, Daniels BG, Polcik M, Verdini A, Floreano L,
Morgante A, Muscat J, Harrison NM, Thornton G, ``Surface to bulk charge
transfer at an alkali metal/metal oxide interface'',
Surface Sci. 547, L859-L864 (2003).
- 408
-
K. Doll, ``Density-functional study of the adsorption of K on the Ag(111)
surface'',
Phys. Rev B 66, art n. 155421 (2002).
- 409
-
K. Doll, ``Density functional study of the adsorption of K on Cu(111)'',
Eur. Phys. J. B 22, 389-393 (2001).
- 410
-
M. Catti, ``Ab initio predicted metastable TlI-like phase in the B1 to B2
high-pressure transition of CaO'',
Phys. Rev. B 68, art 100101 (2003).
- 411
-
A. Lichanot, M. Rerat, M. Catti, ``Theoretical ab initio calculations of
structure factors of fluorite CaF'',
Acta Crystallogr. A51, 323-328 (1995).
- 412
-
Huisinga M, Puchin VE, Reichling M, ``Photoemission from pure and electron
irradiated CaF'',
Nuclear Instruments & methods in physics research B 141,
528-532 (1998).
- 413
-
Puchina AV, Puchin VE, Huisinga M, Bennewitz R, Reichling M, ``Theoretical
modelling of steps and surface oxidation on CaF2'',
Surf. Sci. 404, 687-691 (1998).
- 414
-
F. Marinelli, P. Masri, A. Lichanot, ``Ab initio calculations of elastic
properties and electronic structure of calcium selenide'',
J. Phys. Chem. of Solids 61, 603-608 (2000).
- 415
-
F. Marinelli, M.P. Habas, A. Lichanot, ``Hartree-Fock and density functional
calculations of the elastic constants of CaO'',
J. Phys. Chem. Solids 62, 661-663 (2001).
- 416
-
El Gridani A; El Mouhtadi M, ``Electronic and structural properties of CaH:
a pseudopotential study'',
J. Mol. Struct.-THEOCHEM 532, 183-193 (2000).
- 417
-
Feng XB, Harrison NM, ``Electronic structure of CaCuO from the B3LYP hybrid
density functional'',
Phys. Rev. B 69, Art. No. 132502 (2004).
- 418
-
C.H. Patterson, ``Charge ordered oxygen ions and bi- and tri-Mn polarons in
La
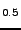 CaMnO'',
CaMnO'',
Mol. Phys. 103, 2507-2512 (2005).
- 419
-
T. Bredow, K. Jug, R.A. Evarestov, ``Electronic and magnetic structure of
ScMnO'',
Phys. Stat. Sol. (b) 243, R10-R12 (2006).
- 420
-
B. Montanari, N.M. Harrison, ``Lattive dynamics of TiOO rutile: influence
of gradient corrections in density functional calculations'',
Chem. Phys. Lett. 364, 528-534 (2002).
- 421
-
J. Muscat, V. Swamy, N.M. Harrison, ``First-principle calculations of the phase
stability of TiO'',
Phys. Rev. B 65, art n. 224112 (2002).
- 422
-
B. Montanari, N.M. Harrison, ``Pressure-induces instabilities in bulk TiO
rutile'',
J. Phys. Cond. Matter 16, 273-292 (2004).
- 423
-
P.J.D. Lindan, J. Muscat, S. Bates, N.M. Harrison, M. Gillan, ``Ab initio
simulation of molecular processes on oxide surfaces'',
Faraday Discussions 106, 135-154 (1997).
- 424
-
L. Giordano, G. Pacchioni, T. Bredow, J. Fernandez Sanz, ``Cu, Ag, and Au atoms
adsorbed on TiO (1 1 0): cluster and periodic calculations.'',
Surface Sci. 471, 21-31 (2001).
- 425
-
R. A. Evarestov, V. P. Smirnov, ``Supercell model of V-doped TiO:
Unrestricted Hartree-Fock calculations'',
Phys. Stat. Sol.(b) 215, 949-956 (1999).
- 426
-
V. A. Veryazov, A. V. Leko, R. A. Evarestov, ``Local characteristics of crystal
electronic structure in the Hartree-Fock method'',
Phys. Solid State 41, 1286-1290 (1999).
- 427
-
W.C. Mackrodt, E.A. Simson and N.M. Harrison, ``An ab initio Hartree-Fock study
of the electron-excess gap states in oxygen-deficient rutile TiO'',
Surf. Sci. 384, 192-200 (1997).
- 428
-
A. Beltran, J. Andres, M. Calatayud, J.B.L. Martins, ``Theoretical study of ZnO
(1 0 0) and Cu/ZnO (1 0 0) surfaces'',
Chem. Phys. Letters 338, 224-230 (2001).
- 429
-
R.A. Evarestov, V.P. Smirnov, ``Supercell model of v-doped TiO:
Unrestricted Hartree-Fock calculations'',
Physica Status Solidi B 215, 949-956 (1999).
- 430
-
Calatayud M, Mori-Sanchez P, Beltran A, Pendas AM, Francisco E, Andres J, Recio
JM, ``Quantum-mechanical analysis of the equation of state of anatase
TiO'',
Phys. Rev. B 64, art no 184113 (2001).
- 431
-
M. Catti and G. Sandrone, ``Ab initio study of corundum-like MeO oxides
(Me=Ti,V,Cr,Fe,Co,Ni)'',
Faraday Discussions 106, 189-203 (1997).
- 432
-
R. A. Evarestov, A. I. Panin, ``Correlation effects in the Unrestricted
Hartree-Fock method for solids: electronic structure of TiO
crystal.'',
Phys. Stat. Sol.(b) 214, R5-R6 (1999).
- 433
-
R. A. Evarestov, A. V. Leko, V. A. Veryazov, ``Unrestricted HF calculations of
crystals with strongly correlated d-electron subsystem: chemical bonding in
TiO crystal'',
Phys. Stat. Sol. (b) 210, R3-R4 (1998).
- 434
-
J. Muscat, N.M. Harrison, G. Thornton, ``Effects of exchange, correlation, and
numerical approximations on the computed properties of the rutile TiO
(100) surface'',
Phys. Rev B 59, 2320-2326 (1999).
- 435
-
Bredow T, Giordano L, Cinquini F, Pacchioni G, ``Electronic properties of
rutile TiO ultrathin films: Odd-even oscillations with the number of
layers'',
Phys. Rev. B 70, 035419 (2004).
- 436
-
R.A. Evarestov, A.V. Bandura, ``Hartree-Fock calculations of electronic
structure of (110)-surface of rutile TiO: comparison of single (2D) and
periodic (3D) slab models'',
Int. J. Quantum Chem. 96, 282-291 (2004).
- 437
-
M.V. Koudriachova, N.M. Harrison, S.W. de Leeuw, ``Density-functional
simlations of lithium intercalation in rutile'',
Phys. Rev. B 65, art n. 235423 (2002).
- 438
-
Zhang YF; Li JQ; Wu LM, ``A density functional study on the electronic
structures of TiX (X = C, N, O). Part II. Investigations on the electronic
structures of TiC bulk and surfaces'',
J. Mol. Struct.-THEOCHEM 531, 1-8 (2000).
- 439
-
Li JQ; Zhang YF; Xiang SC; Chiu YN, ``A density functional study on the
electronic structures of TiX (X = C, N, O). Part I. The electronic structures
and bonding properties of TiN bulk and (001) surface'',
J. Mol. Struct.-THEOCHEM 530, 209-216 (2000).
- 440
-
Li JQ; Zhang YF, ``A density functional study on the electronic structures of
TiN solid'',
Chinese J. of Chemistry 18, 286-293 (2000).
- 441
-
J. Muscat, N.M. Harrison, ``The physical and electronic structure of the rutile
(001) surface'',
Surf. Sci. 446, 119-127 (2000).
- 442
-
P. Persson, S. Lunell, L. Ojamæ, ``Electronic interactions between aromatic
adsorbates and metal oxide substrates calculated from first principles'',
Chem. Phys. Lett. 364, 469-474 (2002).
- 443
-
V. Swamy, J. Muscat, J.D. Gale, N.M. Harrison, ``Simulation of low index rutile
surfaces with a transferable variable-charge Ti-O interatomic potential and
comparison with ab initio results'',
Surface Sci. 504, 115-124 (2002).
- 444
-
L.S. Dubrovinsky, N.A. Dubrovinskaia, V. Swamy, J. Muscat, N.M. Harrison,
R. Ahuja, B. Holm, B. Johansson, ``Cotunnite-structured titanium dioxide: the
hardest known oxide.'',
Nature 410, 653-654 (2001).
- 445
-
Martinsky C; Minot C, ``A theoretical investigation of structure of dinuclear
titanium complexes'',
Surf. Sci. 467, 152-168 (2000).
- 446
-
S. Piskunov, Yu.F. Zhukovskii, E.A. Kotomin, Yu.N. Shunin, ``Hartree-Fock
calculations for a cubic SrTiO crystal'',
Comp. Modelling and New Technologies 4, 7-17 (2000).
- 447
-
E. Heifets, R.I. Eglitis, E.A. Kotomin, J. Maier, G. Borstel,
``First-principles calculations for SrTiO surface structure'',
Surface Sci. 513, 211-220 (2002).
- 448
-
E. Heifets, W.A. Goddard, E.A. Kotomin, R.I. Eglitis, G. Borstel, ``Ab initio
calculations of the SrTiO (110) polar surface.'',
Phys. Rev B 69, 035408 (2004).
- 449
-
G. Borstel, R.I. Eglitis, E.A. Kotomin, E. Heifets, ``Modelling of defects and
surfaces in perovskite ferroelectrics'',
Physica Status Solidi B 236, 253-264 (2003).
- 450
-
S. Piskunov, E. Heifets, R.I. Eglitis, G. Borstel, ``Bulk properties and
electronic structure of SrTiO, BaTiO, PbTiO perovskites: an ab
initio HF/DFT study'',
Comp. Materials Sci. 29, 165-178 (2004).
- 451
-
J. Carrasco, F. Illas, N. Lopez, E.A. Kotomin, Yu.F. Zhukovskii, S. Piskunov,
J. Maier, K. Hermansson, ``First principles simulations of F centers in cubic
SrTiO'',
Physica Status Solidi (c) 2, 153-158 (2005).
- 452
-
R.A. Evarestov, E. A. Kotomin, Yu. F. Zhukovskii, ``DFT study of a single F
center in cubic SrTiO perovskite'',
Int. J. Quantum Chem. 106, 2173-2183 (2006).
- 453
-
J. Carrasco, F. Illas, N. Lopez, E.A. Kotomin,Yu. F. Zhukovskii, R.A.
Evarestov, Yu. A. Mastrikov, S. Piskunov and J. Maier, ``First-principles
calculations of the atomic and electronic structure of F centers in the bulk
and on the (001) surface of SrTiO'',
Phys. Rev. B 73, Art. N. 064106 (2006).
- 454
-
F. Corà, ``The performance of hybrid density functionals in solid state
chemistry: the case of BaTiO'',
Mol. Phys. 103, 2483-2496 (2005).
- 455
-
Fuks D, Dorfman S, Piskunov S, Kotomin EA, ``Ab initio thermodynamics of
BacSr(1-c)TiO3 solid solutions'',
Phys. Rev. B 71, Art. N. 01411 (2005).
- 456
-
de Lazaro RC, Longo E, Beltran A, Sambrano JR, ``Structural and electronic
properties of PbTiO3: Density functional theory applied to periodic
models.'',
Quimica Nova 28, 10-18 (2005).
- 457
-
S. Piskunov, E.A. Kotomin, E. Heifets, J. Maier, R.I. Eglitis, G. Borstel,
``Hybrid DFT calculations of the atomic and electronic structure for ABO
perovskite (001) surfaces'',
Surface Sci. 575, 75-88 (2005).
- 458
-
R.I. Eglitis, S. Piskunov, E. Heifets, E.A. Kotomin, G. Borstel, ``Ab initio
study of the SrTiO, BaTiO, PbTiO (001) surfaces'',
Ceramics Int. 30, 1989-1992 (2004).
- 459
-
R.A. Evarestov, S. Piskunov, E.A. Kotomin, G. Borstel, ``Single impurities in
insulators: Ab initio study of Fe-doped SrTiO'',
Phys. Rev. B 67, 064101 (2003).
- 460
-
Fédéric Labat, Philippe Baranek, Christophe Domain, Christian Minot, and
Carlo Adamo, ``Density functional theory analysis of the structural and
electronic properties of TiO rutile and anatase polytypes: Performances
of different exchange-correlation functionals'',
J. Chem. Phys. 26, Art. N. 154703 (2007).
- 461
-
Yu.F. Zhukovskii, S. Piskunov, E.A. Kotomin, O. Sychev and G. Borstel, ``Ab
initio modeling of copper adhesion on regular BaTiO(0 0 1) surfaces'',
Microelectronic Engineering 81, 467-471 (2005).
- 462
-
K.B. Joshi, B.K. Sharma, ``Electronic structure and momentum density
distribution of titanium dioxide'',
J. Alloys and Compounds 440, 51-56 (2007).
- 463
-
Droubay T, Rosso KM, Heald SM, McCready DE, Wang CM, Chambers SA, ``Structure,
magnetism, and conductivity in epitaxial Ti-doped alpha-FeO hematite:
Experiment and density functional theory calculations'',
Phys. Rev. B 75, Art. N. 104412 (2007).
- 464
-
J.Y. Kempf, B. Silvi, A. Dietrich, C.R.A. Catlow, B. Maigret, ``Theoretical
investigations of the electronic properties of Vanadium oxides. 1.
Pseudopotential periodic Hartree-Fcok study of VO crystal lattice'',
Chem. Mater. 5, 641-647 (1993).
- 465
-
S.V. Vybishchiko, J. Sauer, ``VO)
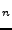 gas-phase clusters (n=1-12)
compared to VO crystal: DFT calculations'',
gas-phase clusters (n=1-12)
compared to VO crystal: DFT calculations'',
J. Phys. Chem. A 105, 8588-8598 (2001).
- 466
-
M. Catti, F. Freyria Fava, C. Zicovich and R. Dovesi, ``High-pressure
decomposition of MCrO spinels (M=Mg, Mn, Zn) by ab initio methods'',
Phys. Chem. Minerals 26, 389-395 (1999).
- 467
-
N.M. Harrison, B.G. Searle, E.A. Seddon, ``An ab initio study of the magnetic
coupling in bimetallic Cr-III cyanides'',
Chem. Phys. Letters 266, 507-511 (1997).
- 468
-
Rehbein C, Michel F, Harrison NM, Wander A, ``Ab initio total energy studies of
the alpha-CrO (0001) and (01(-1)2) surfaces'',
Surf. Rev. Letters 5, 337-340 (1998).
- 469
-
Rehbein C, Harrison NM, Wander A, ``The structure of the Alpha-CrO
(0001) surface: an ab initio total energy study'',
Phys. Rev. B 54, 14066-14070 (1996).
- 470
-
A.V. Olenev, O.S. Oleneva, M. Lindsjö , L.A. Kloo, A.V. Shevelkov,
``Coordinated and clathrated guests in the
 HgAs]
HgAs] bicompartmental framework: synthesis, crystal and electronic structure, and
properties of the novel supramolecular complexes [HgAs](CrBr)Br
and [HgAs](FeBr)Hg
bicompartmental framework: synthesis, crystal and electronic structure, and
properties of the novel supramolecular complexes [HgAs](CrBr)Br
and [HgAs](FeBr)Hg '',
'',
Chem. Eur. J. 9, 3201-3208 (2003).
- 471
-
Waskowska A, Gerward L, Olsen JS, Feliz M, Llusar R, Gracia L, Marques M, Recio
JM, ``High-pressure behaviour of selenium-based spinels and related
structures - an experimental and theoretical study'',
J. Phys.-Condens. Mat 16, 53-63 (2004).
- 472
-
W.C. Mackrodt, E.A. Williamson, D. Williams and N.L. Allan, ``A first
principles Hartree-Fock description of MnO at high pressures'',
Philos. Mag B 77, 1063-1075 (1998).
- 473
-
W.C. Mackrodt and E.A Simson, ``Cation valence charge states of MnFeO -
An ab initio Hartree-Fock study'',
J. Chem. Soc. Faraday Transactions 92, 2043-2047
(1996).
- 474
-
Nicastro M; Kuzmin M; Patterson CH, ``Spin and orbital ordering in CaMnO
and LaMnO: UHF calculations and the Goodenough model'',
Comp. Mat. Science 17, 445-449 (2000).
- 475
-
G. Zheng, C.H. Patterson, ``Ferromagnetic polarons in
LaCaMnO and La
 Ca
Ca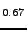 MnO'',
MnO'',
Phys. Rev B 67, 220404 (2003).
- 476
-
R. Tappero, Ph D'Arco and A. Lichanot, ``Electronic structure of alpha-MnS
(alabandite): An ab initio study'',
Chem. Phys. Letters 273, 83-90 (1997).
- 477
-
R.I. Hines, N.L. Allan, G.S. Bell, W.C. Mackrodt, ``An ab initio Hartree-Fock
study of the magnetic states of the polymorphs of MnS'',
J. Phys. Cond. Matter 9, 7105-7118 (1997).
- 478
-
W.C. Mackrodt and E.A. Williamson, ``First-principles description of the
valence charge states in rutile MnO'',
J. Chem. Soc. Faraday Trans. 93, 3295-3300 (1997).
- 479
-
A. Chartier, P. D'Arco, R. Dovesi and V.R. Saunders, ``An ab initio
Hartree-Fock investigation of the structural, electronic and magnetic
properties of MnO-Hausmannite'',
Phys. Rev. B 60, 14042-14048 (1999).
- 480
-
W.C. Mackrodt and E.A. Williamson, ``First principles description of the
valence states in manganese oxides'',
Ber. Bunsenges. Phys. Chem. Chem. Phys. 101, 1215-1221
(1997).
- 481
-
Usvyat DE, Evarestov RA, Smirnov VP, ``Wannier functions and chemical bonding
in crystals with the perovskite-like structure: SrTiO3, BaTiO3, PbTiO3, and
LaMnO3'',
Int. J. Quantum Chem. 100, 352-359 (2004).
- 482
-
Y.S. Su, T.A. Kaplan, S.D. Mahanti, J.F. Harrison, ``Electronic structure of
LaMnO3 in the ab initio crystal Hartree-Fock approximation'',
Phys. Rev. B 61, 1324-1329 (2000).
- 483
-
R.A. Evarestov, E.A. Kotomin, E. Heifets, J. Maier, G. Borstel, ``Ab initio
Hartree-Fock calculations of LaMnO (110) surfaces'',
Solid State Comm. 127, 367-371 (2003).
- 484
-
D. Munoz, N.M. Harrison, F. Illas, ``Electronic and magnetic structure of
LaMnO from hybrid density functional theory'',
Phys. Rev. B 69, art. n. 085115 (2004).
- 485
-
K. Doll, R. Dovesi, R. Orlando, ``Analytical Hartree-Fock gradients with
respect to the cell parameter for systems periodic in three dimensions'',
Theor. Chem. Acc. 112, 394-402 (2004).
- 486
-
Becker U, Hochella MF, Aprà E, ``The electronic structure of hematite001
surfaces: Applications to the interpretation of STM images and heterogeneous
surface reactions'',
American Mineralogist 81, 1301-1314 (1996).
- 487
-
W.C. Mackrodt, ``The nature of valence band holes in pure and Fe-doped NiO: An
ab initio Hartree-Fock study'',
Ber. Bunsenges. Phys. Chem. Chem. Phys. 101, 169-175
(1997).
- 488
-
Aray Y, Rodriguez J, ``Study of CO adsorption on the FeO adsorption on
Laplacian of the electronic charge density'',
Surf. Sci. 405, L532-L541 (1998).
- 489
-
Ahdjoudj J, Martinsky C, Minot C, VanHove MA, Somorjai GA, ``Theoretical study
of the termination of the FeO (111) surface'',
Surface Sci. 443, 133-153 (1999).
- 490
-
P. Schwerdtfeger, T. Söhnel, M. Pempointner, J.K. Laerdahl, F.E. Wagner,
``Comparison of ab initio and density functional calculations of
electric field gradients: The
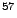 Fe nuclear quadrupole moment from
Mössbauer data'',
Fe nuclear quadrupole moment from
Mössbauer data'',
J. Chem. Phys. 115, 5913-5924 (2001).
- 491
-
Kreitlow J, Menzel D, Wolter AUB, Schoenes J, Sullow S, Feyerherm R, Doll K,
``Pressure dependence of CNH-mediated superexchange in
XCl(CNH)(2) (X=Fe,Co,Ni)'',
Phys. Rev. B 72, Art. No. 134418 (2005).
- 492
-
Feyerherm R, Loose A, Ishida T, Nogami T, Kreitlow J, Baabe D, Litterst FJ,
Sullow S, Klauss HH, Doll K, ``Weak ferromagnetism with very large canting in
a chiral lattice: Fe(pyrimidine)Cl'',
Phys. Rev. B 69 (2004).
- 493
-
Grau-Crespo R, Cora F, Sokol AA, de Leeuw NH, Catlow CRA, ``Electronic
structure and magnetic coupling in FeSbO: A DFT study using hybrid
functionals and GGA+U methods'',
Phys. Rev. B 73, Art. N. 035116 (2006).
- 494
-
N.L. Ross, G.V. Gibbs, K.M. Rosso, ``Potential docking sites and positions of
hydrogen in high pressure silicates'',
American Mineralogist 88, 1452-1459 (2003).
- 495
-
C.H. Patterson, ``Role of defects in ferromagnetism in Zn
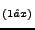 CoO: A
hybrid density-functional study'',
CoO: A
hybrid density-functional study'',
Phys. Rev B 74, Art. n. 144432 (2006).
- 496
-
R. Weihrich, I. Anusca, ``Half Antiperovskites III: crystallographic and
electronic structure effects in Co-Shandites CoSn-xInxS2.'',
Z. Anorg. Allg. Chem. 632, 1531-1537 (2006).
- 497
-
R. Dovesi, C. Pisani and C. Roetti, ``Calculations of nickel band structure and
adsorption on nickel thin films, using an extension of the CNDO formalism'',
Surf. Sci. 103, 482-495 (1981).
- 498
-
Mackrodt WC; Noguera C, ``Bulk and (100) surface d -d excitation energies in
NiO from first-principles Hartree-Fock calculations'',
Surf. Sci. 457, L386-L390 (2000).
- 499
-
Noguera C; Mackrodt WC, ``Ab initio study of ground and excited states of
NiO(100) monolayer'',
J. Phys. - Cond. Matter 12, 2163-2181 (2000).
- 500
-
D. S. Middlemiss and W. C. Mackrodt, ``First principles study of d→d
excitations in bulk NiO'',
Mol. Phys. 103, 2513-2525 (2005).
- 501
-
X.B. Feng, N.M. Harrison, ``Metal-insulator and magnetic transition of NiO at
high pressures'',
Phys. Rev. B 69, art n. 035114 (2004).
- 502
-
M.D. Towler, N.L. Allan, N.M. Harrison, V.R. Saunders, W.C. Mackrodt and E.
Aprà, ``Ab initio study of MnO and NiO'',
Phys. Rev. B 50, 5041-5054 (1994).
- 503
-
S. Casassa, A.M. Ferrari, M. Busso, C. Pisani, ``Structural, electronic and
magnetic properties of the NiO monolayer epitaxially grown on the (001) Ag
surface: an ab-initio density functional study.'',
J. Phys. Chem. B 106, 12978-12985 (2002).
- 504
-
F. Cinquini, L. Giordano, G. Pacchioni, A.M. Ferrari, C. Pisani, C. Roetti,
``Electronic structure of NiO/Ag(100) thin films from DFT+U and hybrid
functional DFT approaches'',
Phys. Rev B 74, Art. n. 165403 (2006).
- 505
-
C.H. Patterson, ``Comparison of hybrid density functional, Hartree-Fock and GW
calculations on NiO'',
Int. J. Quantum Chem. 106, 3383-3386 (2006).
- 506
-
Su Yen-Sheng, T.A. Kaplan, S.D. Mahanti and J.F. Harrison, ``Crystal
Hartree-Fock calculations for LaNiO and La CuO'',
Phys. Rev B 59, 10521-10529 (1999).
- 507
-
G.S. Chandler, B.N. Figgis, Zucheng Li, ``Ab initio calculation of
experimental structure factors for Ni(NH)(NO)'',
Phys. Chem. Chem. Phys. 2, 3743-3751 (2000).
- 508
-
Agnoli S, Sambi M, Granozzi G, Schoiswohl J, Surnev S, Netzer FP, Ferrero M,
Ferrari AM, Pisani C, ``Experimental and theoretical study of a surface
stabilized monolayer phase of nickel oxide on Pd(100)'',
J. Phys. Chem. B 109, 17197-17204 (2005).
- 509
-
Wang, Wenfeng; Li, Junqian; Zhang, Yongfan, ``The orbital interaction of
adsorbed CO on NiO (001;111) surface: A periodic density functional theory
study'',
Applied Surface Sci. 252, 2673-2683 (2006).
- 510
-
R. Weihrich, I. Anusca, M. Zabel, ``Halbantiperovskites: on the crystal
structure of the shandites (NiInS) and their structure relations
to HgSCl and KSnO.'',
Anorg. Allg. Chem. 631, 1463-1470 (2005).
- 511
-
E. Ruiz, M. Llunell, P. Alemany, ``Calculation of exchange coupling constants
in solid state transition metal compounds using localized atomic orbital
basis sets'',
J. Solid State Chem. 176, 400-411 (2003).
- 512
-
T. Bredow, G. Pacchioni, ``Comparative periodic and cluster ab initio study on
CuO(111)/CO'',
Surf. Sci. 373, 21-32 (1997).
- 513
-
Buljan A, Alemany P, Ruiz E, ``Electronic structure and bonding in CuMO2 (M =
Al, Ga, Y) delafossite-type oxides: An ab initio study'',
J. Phys. Chem. B 103, 8060-8066 (1999).
- 514
-
J.K. Perry, J. Tahir-kheli, W.A. Goddard III, ``Antiferromagnetic band
structure of LaCuO: Becke-3-Lee-Yang-Parr calculations'',
Phys. Rev. B 63, 144510 (2001).
- 515
-
J.K. Perry, J. Tahir-kheli, W.A. Goddard III, ``Ab initio evidence for the
formation of impurity d(3z)(2)-(2)(r) holes in doped
La
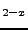 SrCuO'',
SrCuO'',
Phys. Rev. B 65, 144501 (2002).
- 516
-
D. Dominguez-Ariza, C. Sousa, N.M. Harrison, M.V. Ganduglia-Piovano, F. Illas,
``Effect of the surface model on the theoretical description of atomic
hydrogen on Cu(001)'',
Surface Sci. 522, 185-197 (2003).
- 517
-
Neef M, Doll K, ``CO adsorption on the Cu(111) surface: A density functional
study'',
Surface Sci. 600, 1085-1092 (2006).
- 518
-
K. Doll, ``Calculation of the work function with a local basis set'',
Surface Sci. 600, L321-L325 (2006).
- 519
-
T. Söhnel, H. Hermann, P. Schwerdtfeger, ``Solid state density functional
calculations for the group 11 monohalides'',
J. Phys. Chem. B 109, 526-531 (2005).
- 520
-
Ruiz E, Llunell M, Cano J, Rabu P, Drillon M, Massobrio C, ``Theoretical
determination of multiple exchange couplings and magnetic susceptibility data
in inorganic solids: The prototypical case of Cu(OH)NO'',
J. Phys. Chem. B 110, 115-118 (2006).
- 521
-
J.E. Jaffe and A.C. Hess, ``Hartree-Fock study of phase changes in ZnO at high
pressure.'',
Phys. Rev.B 48, 7903 (1993).
- 522
-
A. Wander, N.M. Harrison, ``Ab initio study of ZnO (100)'',
Surface Sci. 457, L342-L346 (2000).
- 523
-
A. Wander, N.M. Harrison, ``Ab initio study of ZnO (11
 0)'',
0)'',
Surface Sci. 468, L851-L855 (2000).
- 524
-
Stahn J, Pucher A, Geue T, Daniel A, Pietsch U, ``Electric-field-induced
electron density response of GaAs and ZnS'',
Europhysics Letters 44, 714-720 (1998).
- 525
-
P. Persson, L. Ojamae, ``Periodic Hartree-Fock study of the adsorption of
formic acid on ZnO((100)'',
Chem. Phys. Lett. 321, 302-308 (2000).
- 526
-
A. Wander, F. Schedin, P. Steadman, A. Norris, R. McGrath, T.S. Turner,
G. Thornton, N.M. Harrison, ``The stability of polar oxide surfaces'',
Phys. Rev. Lett 86, 3811-3814 (2001).
- 527
-
Davaasambuu J; Daniel A; Stahn J; Pietsch U, ``Harmonic and anharmonic thermal
vibrations in cubic ZnSe'',
Z. Fuer Kristallographie 216, 22-25 (2001).
- 528
-
Franco R, Mori-Sanchez P, Recio JM, Pandey R, ``Theoretical compressibilities
of high-pressure ZnTe polymorphs'',
Phys. Rev. B 68, art no 195208 (2003).
- 529
-
Broadley S, Gal ZA, Cora F, Smura CF, Clarke SJ, ``Vertex-linked ZnOS
tetrahedra in the oxysulfide BaZnOS: a new coordination environment for zinc
in a condensed solid'',
Inorganic Chemistry 44, 9092-9096 (2005).
- 530
-
V.M. Bermudez, ``Theoretical study of hydrogen adsorption on the GaN(0 0 0 1)
surface'',
Surface Sci 565, 89-102 (2004).
- 531
-
C. Molteni, L. Miglio, L. Colombo and M. Causà, ``Transferable
tight-binding potentials for molecular dynamics simulations in GaAs'',
Vuoto 22, 105-106 (1994).
- 532
-
C. Gatti, F. Cargnoni, L. Bertini, ``Chemical information from the source
function.'',
J. Comp. Chem. 24, 422-436 (2003).
- 533
-
H. He, R. Orlando, M. A. Blanco and R. Pandey, ``First principles study of the
structural, electronic and optical properties in GaO in its
monoclinic and hexagonal phases'',
Phys. Rev. B 74, Art. n. 195123 (2006).
- 534
-
R. Pandey, M. Rerat, M. Causá, ``First-principles study of stability, band
structure and optical properties of the ordered Ge 0.50 Sn 0.50 alloy'',
Appl. Phys. Letters 75, 4127-4129 (1999).
- 535
-
Pandey R; Rerat M; Darrigan C; Causa M, ``A theoretical study of stability,
electronic, and optical properties of GeC and SnC'',
J. Applied Phys. 88, 6462-6466 (2000).
- 536
-
Marinelli F; Dupin H; Lichanot A, ``Comparison of elastic constants and
electronic structures in the series of the alkaline-earth selenides: a
quantum chemical approach'',
J. Phys. Chem. Solids 61, 1707-1715 (2000).
- 537
-
A.V. Mudring, J. Nuss, U. Wedig, M. Jansen, ``Mixed valent gold oxides:
syntheses, structures, and properties of RbAuO,
RbAuO, CsAuO'',
J. Solid State Chem. 155, 29-36 (2000).
- 538
-
El Gridani AF; El Bouzaidi RD; El Mouhtadi M, ``Elastic, electronic and crystal
structure of SrH by the method of pseudopotentials'',
J. Mol. Struct.-THEOCHEM 531, 193-210 (2000).
- 539
-
Gunhold A, Beuermann L, Gomann K, Borchardt G, Kempter V, Maus-Friedrichs W,
Piskunov S, Kotomin EA, Dorfman S, ``Study of the electronic and atomic
structure of thermally treated SrTiO3(110) surfaces'',
Surf. Interface Anal. 35, 998-1003 (2003).
- 540
-
M.Merawa, B. Civalleri, P. Ugliengo, Y. Noel and A. Lichanot, ``Structural,
electronic and vibrational properties of solid Sr(OH), calculated with
different hamiltonians'',
J. Chem. Phys. 119, 1045-1052 (2003).
- 541
-
Gennard S, Cora F, Catlow CRA, ``Comparison of the bulk and surface properties
of ceria and zirconia by ab initio investigations'',
J. Phys. Chem. B 103, 10158-10170 (1999).
- 542
-
T. Bredow, M. Lerch, ``Anion distribution in ZrON'',
ZEITSCHRIFT FUR ANORGANISCHE UND ALLGEMEINE CHEMIE 630,
2262-2266 (2004).
- 543
-
Ph. Baranek, C.M. Zicovich-Wilson, C. Roetti, R. Orlando, R. Dovesi, ``Well
localized Crystalline Orbitals as obtained from Bloch functions. The case of
KNbO'',
Phys. Rev. B 64, 125102 (2001).
- 544
-
Fu L, Yaschenko E, Resca L, Resta R, ``Polarization properties of KNbO:
comparison between Hartree- Fock and density-functional calculations'',
Solid State Comm. 112, 465-470 (1999).
- 545
-
Ph. Baranek, R. Dovesi, ``Ab initio approach to the ferroelectric properties of
ABO perovskites: the case of KNbO'',
Ferroelectrics 268, 155-162 (2002).
- 546
-
F. Corà, A. Patel, N.M. Harrison, C. Roetti and C.R.A. Catlow, ``An ab-initio
Hartree-Fock study of -MoO'',
J. Mater. Chem. 7, 959-967 (1997).
- 547
-
V. Alexiev, R. Prins, T. Weber, ``DFT study of MoS and hydrogen adsorbed on
the (10
 0) face'',
0) face'',
Phys. Chem. Chem. Phys. 3, 5326-5336 (2001).
- 548
-
F. Frechard, P. Sautet, ``Hartree-Fock ab-initio study of the geometric and
electronic structure of RuS and its (100) and (111) surfaces.'',
Surf. Sci. 336, 149-165 (1995).
- 549
-
J.R.B. Gomes, F. Illas, N. Cruz Hernandez, J.F. Sanz, A. Wander, N.M. Harrison,
``Surface model and exchange-correlation functional effects on the
description of Pd/-AlO(0001).'',
J. Chem. Phys. 116, 1684-1691 (2002).
- 550
-
K. Doll, P. Pyykko, H. Stoll, ``Closed-shell interaction in silver and gold
chlorides'',
J. Chem. Phys. 109, 2339-2345 (1998).
- 551
-
K. Doll and N.M. Harrison, ``Theoretical study of chlorine adsorption on the
Ag(111) surface'',
Phys. Rev. B 63, n.165410 (2001).
- 552
-
M. Catti, ``Kinetic mechanisms of the pressure-driven phase transitions of
AgI'',
Phys. Rev B 72, Art. N. 06415 (2005).
- 553
-
Y Dou, R.G. Egdell, D.S.L. Law, N.M Harrison, B.G. Searle, ``An experimental
and theoretical investigation of the electronic structure of CdO'',
J. Phys. Cond. Matter 10, 8447-8458 (1998).
- 554
-
M. Calatayud, J. Andres, A. Beltran, ``A theoretical analysis of adsorption and
dissociation of CHOH on the stoichiometric SnO surface.'',
Surf. Sci. 430, 213-222 (1999).
- 555
-
Melle-Franco M; Pacchioni G, ``CO adsorption on SnO2(110): cluster and periodic
ab initio calculations'',
Surf. Sci. 461, 54-66 (2000).
- 556
-
T. Bredow, G. Pacchioni, ``NO adsorption on the stoichiometric and reduced
SnO(110) surface'',
Theoretical Chemistry Accounts 114, 52-59 (2005).
- 557
-
C. A. Stückl, F. Rau, R. Weihrich, ``Charge transfer and chemical hardness
along a substitution path inmetastable Au-Sb alloys'',
Z. Anorg. Allg. Chem. 629, 1812-1824 (2003).
- 558
-
B. ellman, ``Ab initio study of the electronic structure of the crystalline
high-mobility organic semiconductor 1,4-diiodobenzene'',
J. Chem. Phys. 125, art n. 074702 (2006).
- 559
-
A. Karpov, J. Nuss, U. Wedig, M. Jansen, ``CsPt: A platinide (-II)
exhibiting complete charge separation'',
Angew. Chem. Int. Ed. 42, 4818-4821 (2003).
- 560
-
H.T. Jiang, R. Pandey, C. Darrigan, M. Rerat, ``First-principle study of
structural, electronic and optical properties of BaF it its cubic,
orthorhombic, and hexagonal phases.'',
J. Phys. Cond. Matter 15, 709-718 (2003).
- 561
-
J. Yang, M. Dolg, ``Valence basis sets for lanthanide 4f-in-core
pseudopotentials adapted for crystal orbital ab initio calculations'',
Theor. Chem. Accounts 113, 212-224 (2005).
- 562
-
S.E. Hill, C.R.A. Catlow, ``A Hartree-Fock periodic study of bulk Ceria'',
Phys. Chem. Solids 54, 411-419 (1993).
- 563
-
C. Mueller, C. Freysoldt, M. Baudin, K. Hermansson, ``An ab initio study of CO
adsorption on ceria (110)'',
Chem. Phys. 318, 180-190 (2005).
- 564
-
D. Munoz-Ramo, J. L. Gavartin, and A. L. Shluger, ``Spectroscopic properties of
oxygen vacancies in monoclinic HfO calculated with periodic and embedded
cluster density functional theory'',
Phys. Rev. B 75, Art. N. 205336 (2007).
- 565
-
Bredow T, Lumey MW, Dronskowski R, Schilling H, Pickardt J, Lerch M,
``Structure and stability of TaON polymorphs'',
ZEITSCHRIFT FUR ANORGANISCHE UND ALLGEMEINE CHEMIE 632,
1157-1162 (2006).
- 566
-
A. Kokalj, M. Causá, ``Periodic Density Functional theory study of Pt(111):
surface features of slabs of different thickness'',
J. Phys. Cond. Matter 11, 7463-7480 (1999).
- 567
-
A. Kokalj, A. Lesar, M. Hodoschek, M. Causá, ``Periodic DFT study of the
PT(111): A p(1x1) atomic oxygen interaction with the surface'',
J. Phys. Chem. B 103, 7222-7232 (1999).
- 568
-
K. Doll, ``CO adsorption on the Pt(111) surface: a comparison of a gradient
corrected functional and a hybrid functional'',
Surface Sci. 573, 464-473 (2004).
crystal development
2007-06-06